


Translate this page into:
Nuclear receptor subfamily 4 group a member 1 eases angiotensin II-arose oxidative stress in vascular smooth muscle cell by boosting nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3 transcription

*Corresponding author: Zhiqiang Yan, Central Laboratory, Fengxian District Central Hospital, Southern Medical University, 6600 Nanfeng Road, Fengxian District, Shanghai, China. & The Third School of Clinical Medicine, Southern Medical University, Guangzhou, Guangdong, China. zqyan@sjtu.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Shen L, Li F, Xia K, Zhan L, Zhang D, Yan Z. Nuclear receptor subfamily 4 group a member 1 eases angiotensin II-arose oxidative stress in vascular smooth muscle cell by boosting nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3 transcription. CytoJournal. 2024;21:43. doi: 10.25259/Cytojournal_86_2024
Abstract
Objective:
Hypertension significantly contributes to morbidity and mortality. Nuclear receptor subfamily 4 group a member 1 (Nur77) participates in regulating oxidative stress, but the mechanism in hypertension remains unclear. This study aimed to explore the function of Nur77 in oxidative stress induced by Angiotensin II (Ang II) in vascular smooth muscle cells (VSMCs) in hypertension.
Material and Methods:
First, models of VSMC with Nur77, nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3 (NLRC3) and tumor necrosis factor receptor-associated factor 6 (TRAF6) knockdown or overexpression were constructed using Short Hairpin RNA (Nur77) or pcDNA3.1 vector, respectively. Next, the putative-binding motifs between Nur77 and NLRC3 promoters were detected by dual luciferase assay. We conducted reverse transcription quantitative polymerase chain reaction (qPCR) and Western blot (WB) analysis to detect Nur77, NLRC3, and TRAF6 levels in VSMCs. Then, cell counting kit-8 assay, 5-ethynyl-2’-deoxyuridine assay, wound-healing assay, enzyme-linked immunosorbent assay, and 2’,7’-dichlorofluorescin diacetate were employed to examine the impact of the knockdown or overexpression of Nur77, NLRC3, and TRAF6 on VSMCs treated with Ang II. The assays measured cell viability and proliferation, cell migration, malondialdehyde levels, and reactive oxygen species levels.
Results:
The overexpression of Nur77 repressed cell growth (P < 0.001), migration (P < 0.01), and oxidative stress (P < 0.01) induced by Ang II in VSMCs. Nur77 transcriptionally promoted the expression of NLRC3 (P < 0.001), and the upregulation of NLRC3 suppressed cell proliferation (P < 0.05) and oxidative stress (P < 0.001) mediated by Ang II. Furthermore, NLRC3 negatively regulated the TRAF6/nuclear factor-kappa B (NF-κB) axis activated by Ang II, which resulted in the repression of hyperproliferation of VSMCs (P < 0.01) and oxidative stress (P < 0.001).
Conclusion:
Nur77 suppressed growth and oxidative stress induced by Ang II in VSMCs by promoting NLRC3 transcription, which, further, repressed the TRAF6/NF-κB axis. This understanding provides novel insights into the pathogenesis of hypertension.
Keywords
Hypertension
Nuclear receptor subfamily 4 group A member 1
Nuclear factor-kappa B
Cell migration
Oxidative stress

INTRODUCTION
Hypertension, which is defined as a blood pressure of ≥140/90 mmHg,[1] is a leading contributor to early mortality. It is a gradually progressive chronic condition that impacts over one billion adults (30–79 years old) globally.[2] Hypertension significantly contributes to the increased risk of various brain and cardiovascular diseases.[3,4] Reactive oxygen species (ROS) produced by oxidative stress have been proven to adversely affect cells and then lead to cell death in hypertension.[5] Chronic hypertension results in impaired endothelium-dependent relaxation, increased arterial stiffness, enhanced contractility, inflammation, vascular calcification, and remodeling. Oxidative stress has been confirmed to aggravate these dysfunctions.[6] Angiotensin II (Ang II) was reported as one of the ROS inducers in vascular cells.[7] A study found that ROS was involved in endothelial function and the regulation of vascular tone in the vascular system; increased ROS production or weakened antioxidant defense can lead to dysfunction in vascular smooth muscle cells (VSMCs), which can result in the progression of hypertension.[8] In VSMCs, ROS induced by Ang II can lead to changes of vascular reactivity, growth, calcification, and fibrosis, which affect vascular remodeling.[9] Characterizing the underlying molecular mechanisms of antioxidant defense to identify potential therapeutic targets is imperative to improve the treatment of hypertension.
Nuclear receptor subfamily 4 group a member 1 (Nur77) plays critical roles in maintaining vascular homeostasis by regulating oxidative stress and vascular remodeling.[10] Nur77 was found to attenuate endothelial dysfunction through activating antioxidant pathways and reducing the production of ROS in vascular endothelium.[11] The knockout of Nur77 increased the ROS production in the aorta of mice.[11] In addition, the expression of Nur77 in pulmonary arteries was significantly reduced in patients with pulmonary arterial hypertension,[12] whereas the overexpression of Nur77 significantly repressed the proliferation and migration of VSMCs.[13] Nur77 also attenuated oxidative stress by inhibiting the phosphorylated recombinant inhibitory subunit of nuclear factor-kappa B alpha (NF-κB) in the cell model of Parkinson’s disease.[14] However, the molecular mechanism of Nur77 in regulating vascular oxidative stress in hypertension remains unclear.
Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3 (NLRC3) is an innate immune sensor that participates in regulating inflammation pathways.[15,16] NLRC3 was found to inhibit the proliferation of artery smooth muscle cells stimulated by platelet-derived growth factor through the phosphatidylinositol 3-kinase/protein kinase B pathway.[17] The NF-κB pathway regulates oxidative stress and is activated in hypertension.[15,18,19] A previous study reported that endothelial tumor necrosis factor receptor-associated factor 6 (TRAF6) deficiency decreased atherosclerosis through repressing the expression of proinflammatory gene downstream of NF-κB and monocyte adhesion.[20] At present, whether NLRC3 is involved in oxidative stress in hypertension through TRAF6/NF-κB is unclear.
Given that the mechanism of Nur77 in hypertensive oxidative stress remains unknown, we analyzed the potential transcriptional targets of Nur77 and explored the crosstalk mechanism among Nur77, NLRC3, and NF-κB in VSMCs. Our study aims to offer new insights into the mechanisms underlying hypertension induced by Ang II and identify potential therapeutic targets.
MATERIAL AND METHODS
Cell culture
Human VSMCs (Cat. #CRL-1999, American Type Culture Collection, identified by mycoplasma and STR) were maintained in F-12K nutrient medium (Cat. #30-2004, ATCC, USA) supplemented with fetal bovine serum (FBS), endothelial cell growth supplement, ascorbic acid (Cat. # A4544, Sigma, USA), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Cat. #H4034, Sigma, USA), transferrin (Cat. # T8158, Sigma, USA), insulin (Cat. # I3536, Sigma, USA), sodium selenite (Cat. #S5261, Sigma, USA), and thermal energy storage (Cat. #V900476, Sigma, USA). VSMCs were treated with Ang II (Cat. #305-23-0101, Sigma, USA) (2 µM, 24 h) to mimic their dynamics during hypertension.[21]
Cell transfection
The overexpression vectors for Nur77 (NM_001202233.2), NLRC3 (NM_178844.4), and TRAF6 (NM_004620.4) were constructed using the pcDNA3.1 vector (Cat. #V79020, Thermo Fisher Scientific, Waltham, Massachusetts, USA) by inserting their full length sequences. The sequences were digested using the corresponding endonuclease, and then, they were connected to the pcDNA3.1 vector. The knockdown vector of Nur77 (5’-TGGTGAAGGAAGTTGTCCGAA-3’) was generated by GenePharm (Shanghai, China). When the cells in the plates reached a confluence level of 80–90%, 4 µg of plasmid DNA (dissolve in 10 µL ddH2O) was mixed with 10 µL of Lipofectamine 2000 (Cat. #11668019, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 250 µL of serum-free medium. The mixture was incubated for 20 min before being added to the wells plated in 6-well plates.
Cell counting kit-8 (CCK-8)
The viability of cells was determined using CCK-8 kits (Cat. #ab228554, Abcam, Cambridge, UK). The cells (1 × 104) were planted and cultured in a 96-well plate for 24 h. Then, 10 µL of CCK-8 solution was added and incubated in the dark for 3 h. The absorbance was measured at λ= 460 nm using a microplate reader (TECAN Spark®, Tecan Group Ltd., Switzerland).
5-Ethynyl-2’-deoxyuridine
Cell growth was determined using the 5-ethynyl-2’-deoxyuridine (EdU) kit (Cat. #ab222421, Abcam, Cambridge, UK). A total of 10 µM of EdU was added to cells at a confluence level of 60–70% for 4 h and then fixed in 4% paraformaldehyde (Cat. #E672002, Sangon, Shanghai, China) before permeabilization. The fixative was discarded, and the cells were washed with phosphate-buffered saline (PBS) 3 times, with 5 min each time. Then, the Apollo staining solution was added into the cell, followed by incubation at room temperature without light. 4’-6-diamidino-2-phenylindole (DAPI, Beyotime, Shanghai, China) was used for nuclear staining. The cells were observed using a fluorescence microscope (Nikon TSR2, Japan). Image J software was employed to quantify the intensity of fluorescence.
Wound-healing assay
The 2.5×106 cells were planted in 12 well plates of 10 mL Dulbecco’s Modified Eagle medium containing 10% fetal bovine serum. When the cells achieved a confluence of 100%, cells in every well were scraped with a 1 mm scraper. After three washes with PBS, the cells were cultured with the serum free medium. After 24 h treatment, the migration distance was recorded and photographed at 0 and 24 h. Image acquisition was conducted using a fluorescent microscope (Nikon TSR2, Japan).
Transwell assay
A total of 0.5 mL of cell suspension (1 × 105/mL) in FBS-free media was planted in migration chambers, with FBS-containing medium (0.5 mL) in the lower well. After incubation for 24 h, the inserts were removed and washed with pre-chilled PBS before fixing the cells in 4% paraformaldehyde and staining in 1% crystal violet (Songon, Shanghai). Any cell presented in the inner section of the inserts was delicately wiped away using a cotton swab. A microscope (Nikon TSR2, Japan) was used to count migration cells on the external surface, and images of these cells were captured.
ROS
ROS was evaluated using 2’,7’-dichlorofluorescin diacetate (DCFH-DA) (Cat. # D6883, Sigma–Aldrich, USA). In brief, after 24 h treatment with Ang II, the medium was changed to serum-free medium with 10 µM of DCFH-DA. Then, it was incubated in dark at 37°C for 30 min. Images were captured using a fluorescent microscope (Nikon TSR2, Japan),[22] Image J software was employed to quantify the intensity of fluorescence. DAPI (Beyotime, Shanghai, China) was used for nuclear staining.
Enzyme-linked immunosorbent assay (ELISA)
Superoxide dismutase (SOD) (Cat. #19160, Sigma, USA), Glutathione peroxidase (GSH-PX) (Cat. #ab193767, Abcam, Cambridge, UK), and malondialdehyde (MDA) (Cat. #ab238537, Abcam, Cambridge, UK) levels in cell lysate were detected by ELISA.
For malondialdehyde (MDA) detection, 50 µL of appropriately diluted bovine serum albumin-conjugated MDA standards or samples were added to the 96-well plate and incubated for 10 min at ambient temperature. Then, the plate was incubated with 50 µL of anti-MDA antibody for 1 h. After the plate was washed 3 times, secondary antibody – horseradish peroxidase (HRP) conjugate (100 µL) was added. The substrate solution was added after three washes. Next, a stop solution was added to the plate followed by the absorbance measured at λ = 450 nm. All samples were assessed in triplicate.
For SOD detection, 100 µL of appropriately diluted standards and samples was added before the addition of 50 µL of HRP conjugate to all wells. Following a 3 h incubation and three washes, tetramethyl benzene (TMB) substrate solution (100 µL) was added and incubated for 10 min. Then, 100 µL of stop solution was added, and the absorbance was measured at λ = 450 nm.
To detect glutathione peroxidase (GSH-PX), 50 µL of appropriately diluted standards and samples were added, followed by adding 50 µL of antibody cocktail. After 1 h incubation and three washes, 100 µL of TMB was added. Then, 100 µL of stop solution was added, and the absorbance was measured at λ = 450 nm.
The total protein concentration was determined using the bicinchoninic acid assay (BCA) (Pierce, Bonn, Germany). The results were presented as the activity or content of SOD, MDA, and GSH-PX per mg protein.
Luciferase assay
The putative-binding motifs between the Nur77 and NLRC3 promoters were analyzed using the JASPAR database (https://jaspar.elixir.no/). The promoter region of NLRC3 was cloned from human VSMCs. Site-directed mutagenesis of the NLRC3 promoter was achieved using a commercial kit (NEB #E0554, Ipswich, MA, USA). A wild-type (WT) or mutant NLRC3 promoter was inserted into the pGL6 reporter vector (Beyotime, Shanghai, China). A luciferase assay kit (Cat. #E1910, Promega, Madison, Wisconsin, USA) was used to evaluate luciferase activity. Co-transfection of the Nur77 overexpression vector with WT/MUT-NLRC3 in VSMCs was performed for 48 h in 96-well plates. Complete medium by itself acted as blank control. The luciferase assay reagent II (100 µL) was added to the cell lysate (20 µL) to record the firefly luciferase activity immediately. Then, Renilla luciferase activity was determined after the addition of Stop and Glo Reagent (100 µL).
Western blot analysis
Total protein was extracted from cells using radioimmunoprecipitation assay buffer added with a protease inhibitor cocktail (Cat. #11697498001, Roche, Basel, Switzerland), and the concentration was measured through BCA (Pierce, Bonn, Germany). Subsequently, proteins (25 µg) were loaded in 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) before transferring to a polyvinylidene difluoride membrane. After blocking for 1 h with 5% skim milk (Cat. #1.15363, Sigma, USA) and incubation at 4°C overnight with indicated primary antibodies, followed by washing with tris buffered saline plus tween-20 (TBST, Cat. #T9039, Sigma, USA), the membrane was incubated with HRP-conjugated secondary antibody (1:2 000, Cat. #58802, CST). A chemiluminescence kit (Cat. #1705060S, BIO-RAD, Hercules, California, USA) was used for blot development. The images were acquired using the ChemicDoc XRS system (Bio-Rad, USA). Image J software was employed to quantify the gray value of bands. The following primary antibodies were used: NLRC3 (1:1000, Cat. #ab77817, Abcam), TRAF6 (1:1000, Cat. #8028, CST, Danvers, Massachusetts, USA), Nur77 (1:1000, Cat. #3960, CST), phosphorylated p65 (Ser536) (1:1000, Cat. #3031, CST), phosphorylated inhibitory subunit of nuclear factor-kappa B alpha (IκBα) (Ser32) (1:1000, Cat. #2859, CST), and glyceraldehyde phosphate dehydrogenase (GAPDH, 1:1000, Cat. #5174, CST).
RNA isolation and real-time quantitative polymerase chain reaction (qPCR)
RNA extraction was conducted using an RNA isolation kit (Cat. # 74104, QIAGEN, Hilden, Germany). Complementary DNAs were prepared using a commercial reverse transcription kit (Cat. #4368814, Applied Biosystems). A Synergetic Binding Reagent (SYBR) Green PCR kit (Cat. #1725270, BIO-RAD, Hercules, CA, United States) was used to conduct qPCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as reference. The expression levels of genes were calculated using the 2−ΔΔCt formula. The sequences of primers for the genes of interest were as follows:
Nur77-F: 5’-ACCCACTTCTCCACACCTTG-3’
Nur77-R: 5’-ACTTGGCGTTTTTCTGCACT-3’
NLRC3-F: 5’-CTGGGAAGGGCAGTCAAG-3’
NLRC3-R: 5’-TGCCTCTGT ATCCTTGAGTC-3’
TRAF6-F: 5’-TCGAACCCTTGAGGACAAAG-3’
TRAF6-R: 5’-CGGGTTTGCCAGTGTAGAAT-3’
GAPDH-F: 5’-CCAGGTGGTCTCCTCTGA-3’
GAPDH-R: 5’-GCTGTAGCCAAATCGTTGT-3’.
Statistical analysis
The experiments were conducted no less than <3 times. Data were shown as mean ± standard deviation. Students’ t-test was performed to assess the significance and between-group differences were assessed for statistical significance. One-way analysis of variance followed with a post hoc test was conducted for multigroup comparisons. GraphPad (GraphPad Software Inc., San Diego, CA) was used to analyze data. P < 0.05 was considered indicative of statistical significance. All experiments were performed with three independent biological replicates and three technical replicates.
RESULTS
Overexpression of Nur77 represses cell growth and oxidative stress mediated by Ang II
We overexpressed Nur77 in human VSMCs to explore the function of Nur77 in cell damage and redox imbalance induced by Ang II. We also observed the mRNA of Nur77 enhanced at 24 h (P < 0.05), 48 h (P < 0.001), and 72 h (P < 0.001) compared with that of the vector group [Figure 1a]. We further detected the protein expression levels of Nur77 significantly increased at 24 h (P < 0.05), 48 h (P < 0.01), and 72 h (P < 0.001) compared with those of the vector group [Figure 1b and c]. The treatment of VSMCs with Ang II promoted cell viability (P < 0.05), while the overexpression of Nur77 suppressed the effect of Ang II (P < 0.001) [Figure 1d]. Furthermore, the overexpression of Nur77 repressed cell proliferation promoted by Ang II (P < 0.001) [Figure 1e and f]. Ang II promoted the migration of VSMCs, while the overexpression of Nur77 inhibited this effect of Ang II on migration (P < 0.01) [Figure 1g-j]. The treatment of Ang II enhanced the level of ROS, while the overexpression of Nur77 reduced ROS elevation induced by Ang II (P < 0.01) [Figure 1k and l]. Similarly, the overexpression of Nur77 repressed the increment in MDA (P < 0.05), the reduction in SOD (P < 0.01), and the decrease in GSH-PX (P < 0.05) induced by Ang II in VSMCs [Figures 1m-o]. Our data suggested that Nur77 repressed the growth and migration induced by Ang II in VSMCs.
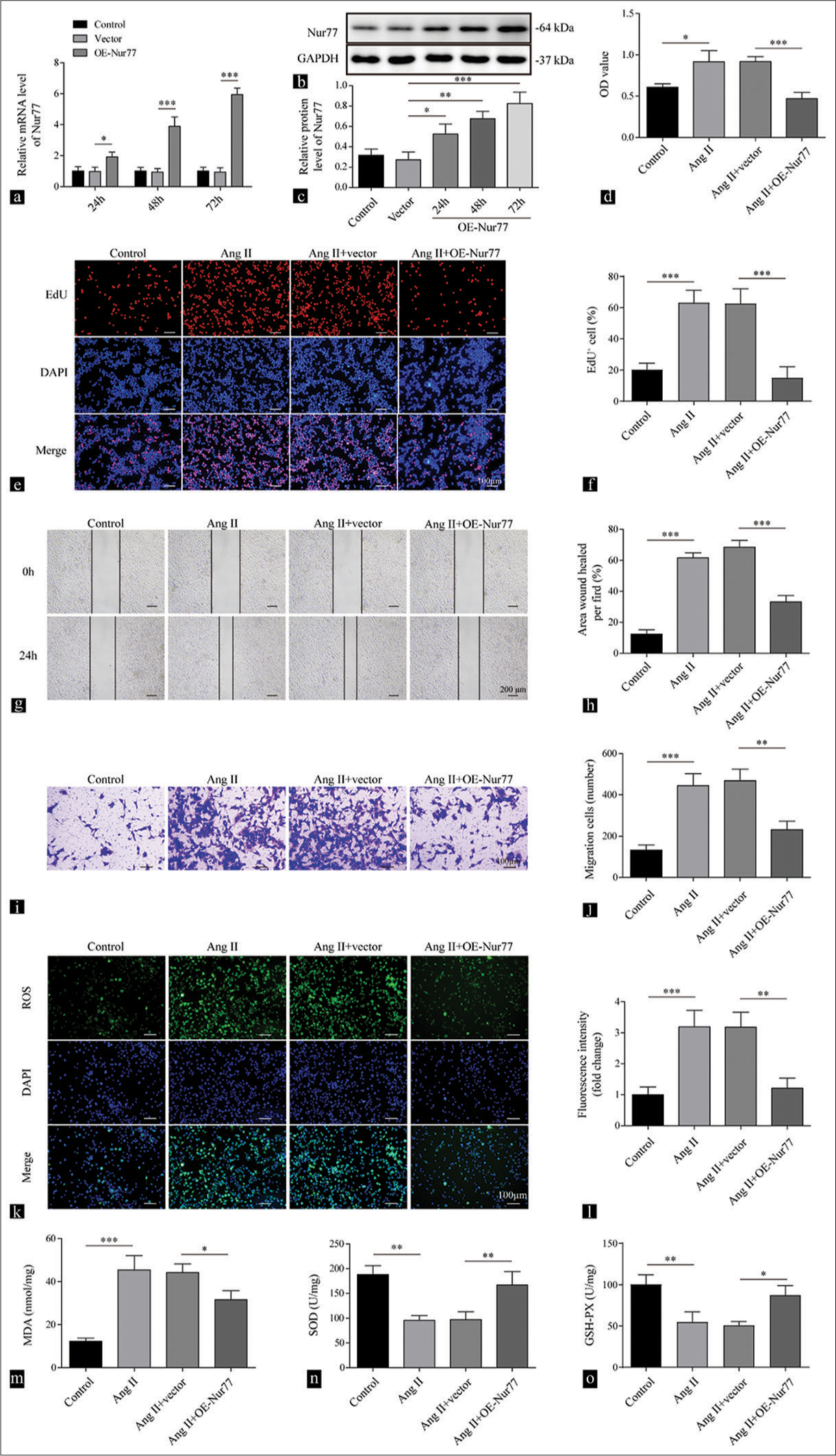
- Overexpression of Nur77 represses cell growth and oxidative stress mediated by Angiotensin II (Ang II). (a-c) Reverse transcription quantitative polymerase chain reaction (a) and Western blot analysis (b and c) were conducted to detect the expression levels of Nur77 at 24, 48, and 72 h in vascular smooth muscle cells (VSMCs) after being treated with vector or oe-Nur77 (n = 3). (d) Cell viability of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-Nur77 were determined by cell counting kit-8 assay (n = 3). (e and f) Proliferation of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-Nur77 was assessed by the EdU assay (n = 3). (g and h) Migration of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-Nur77 at 0 and 24 h was evaluated by the wound-healing assay (n = 3). (i and j) Transwell assay was performed to assess the migration capacity of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) +vector, and Ang II (2 µM) + oe-Nur77 (n = 3). Crystal violet was used to stain migration cell. (k and l) Reactive oxygen species production of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-Nur77 was measured by 2’,7’-dichlorofluorescin diacetate (n = 3). (m-o) Enzyme-linked immunosorbent assay was employed to determine the levels of malondialdehyde, superoxide dismutase, and glutathione peroxidase of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-Nur77 (n = 3). (* P < 0.05, ** P < 0.01, *** P < 0.001. EdU: 5-ethynyl-2’-deoxyuridine, AngII: Angiotensin II, Nur77: Nuclear receptor subfamily 4 group a member 1)
Nur77 transcriptionally promotes the expression of NLRC3
We used the JASPAR software to analyze the potential transcriptional target of Nur77 for determining its molecular mechanism in hypertensive vascular remodeling. The results displayed the binding between Nur77 and the NLRC3 promoter [Figure 2a]. The overexpression of Nur77 enhanced luciferase activity in cells expressing the WT-NLRC3 promoter but did not those expressing the MUT-NLRC3 promoter (P < 0.001) [Figure 2b]. In addition, we knocked down Nur77 in VSMCs using shNur77 (P < 0.01) [Figure 2c-e]. NLRC3 mRNA and protein levels were upregulated after transfection with Nur77 overexpression (P < 0.001) [Figure 2f-h]. The knockdown of Nur77 reduced the mRNA (P < 0.05) and protein (P < 0.01) expression of NLRC3 [Figure 2f-h]. These findings indicated that Nur77 promoted the transcription of NLRC3 and upregulated the expression of NLRC3.
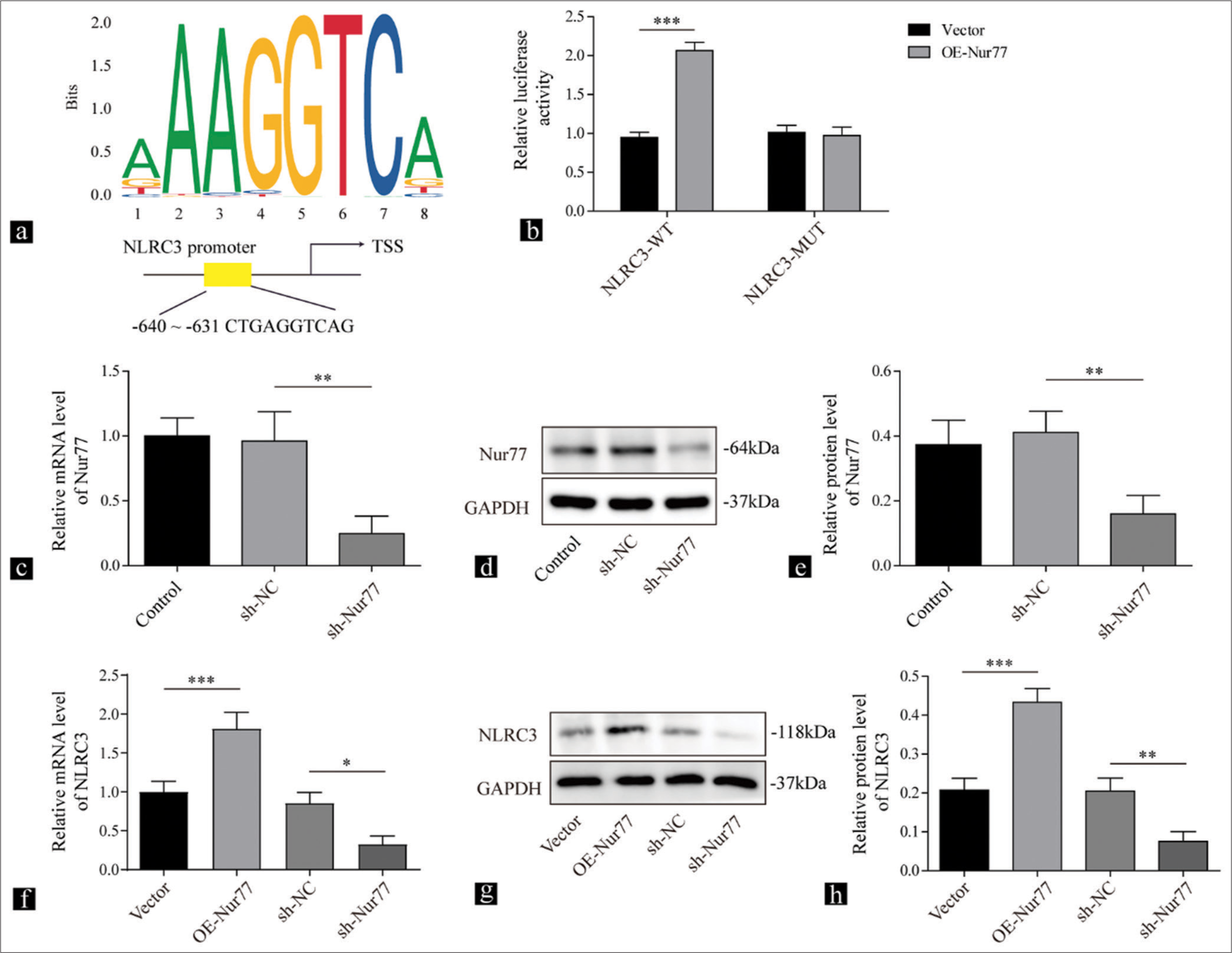
- Nur77 transcriptionally promotes the expression of NLRC3. (a) Jaspar Database of Transcription Factor Binding Site (JASPAR) predictions for putative Nur77 binding region in the NLRC3 promoter. (b) Relative luciferase activity of NLRC3 reporter in vascular smooth muscle cells (VSMCs) transfected with vector or Nur77 evaluated by dual-luciferase assay (n = 3). (c) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) and (d and e) Western blot analysis were conducted to detect the expression levels of Nur77 in VSMCs after being treated with sh-NC or sh-Nur77 (n = 3). (f) RT-qPCR and (g and h) Western blot analysis were performed to determine the expression of NLRC3 in VSMCs transfected with oe-Nur77, sh-NC, or sh-Nur77 (n = 3). (* P < 0.05, ** P < 0.01, *** P < 0.001. AngII: Angiotensin II, Nur77: Nuclear receptor subfamily 4 group a member 1, NLRC3: Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3.)
Upregulation of NLRC3 suppresses cell growth and oxidative stress dependent on Ang II
We overexpressed NLRC3 in VSMCs to explore the potential involvement of NLRC3 in regulating oxidative stress induced by
Ang II in VSMCs. The transfection of VSMCs with the NLRC3 overexpression plasmid increased the mRNA (P < 0.001) and protein (P < 0.01) expression of NLRC3 [Figure 3a-c]. The upregulation of NLRC3 significantly reversed cell viability (P < 0.05) and proliferation (P < 0.001) induced by Ang II [Figure 3d-f]. In addition, the overexpression of NLRC3 suppressed cell migration (P < 0.05) induced by Ang II in VSMCs [Figure 3g-j]. Moreover, the overexpression of NLRC3 reduced the level of ROS in VSMCs treated with Ang II (P < 0.001) [Figure 3k and l]. Similarly, the increase in MDA (P < 0.001), the reduction in SOD (P < 0.05), and the decline in GSH-PX (P < 0.001) caused by Ang II treatment was repressed by the overexpression of NLRC3 in VSMCs [Figure 3m-o]. Collectively, these data demonstrated that NLRC3 counteracted the pro-growth effect and ROS induction by Ang II.
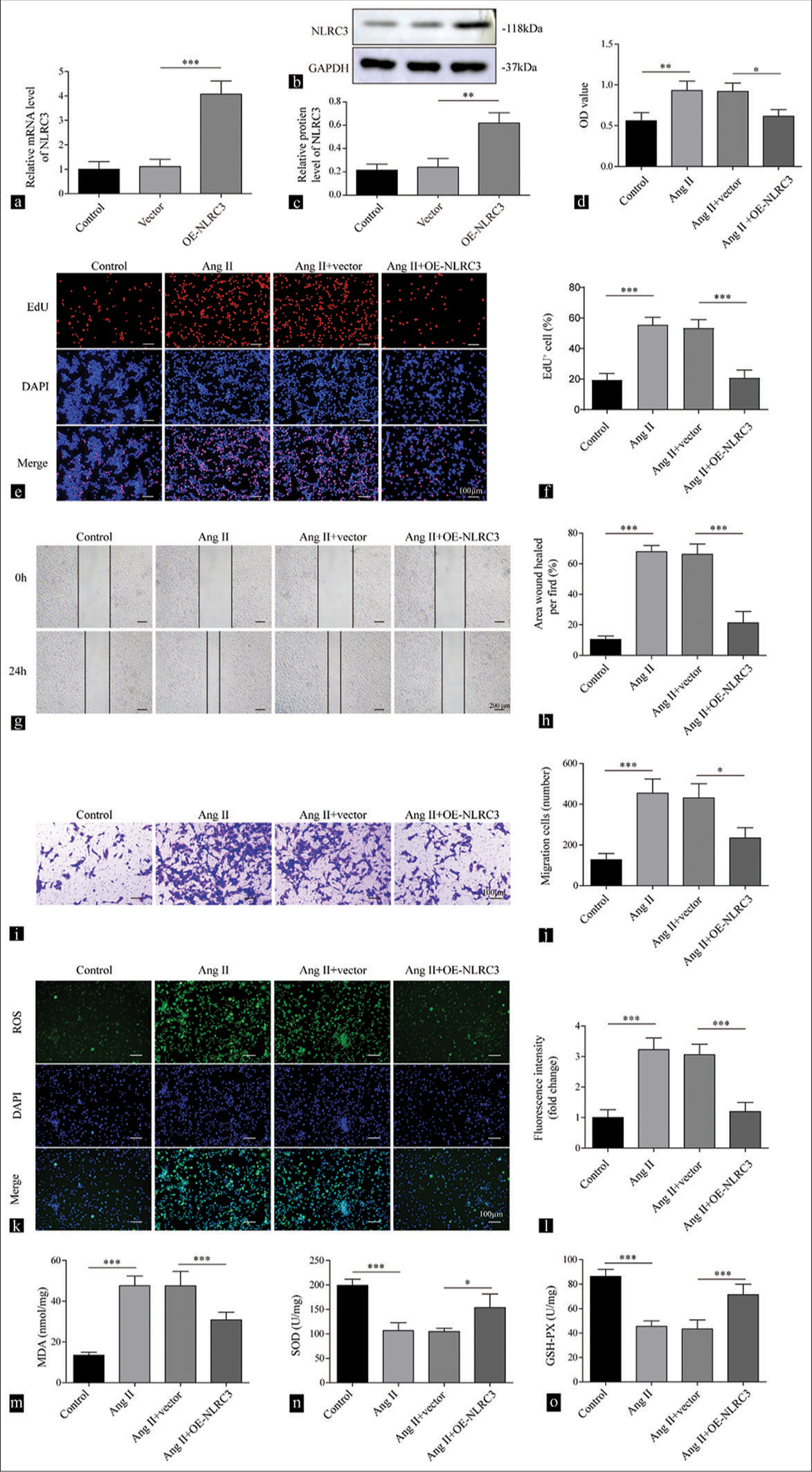
- Upregulation of NLRC3 suppresses cell proliferation and oxidative stress mediated by Angiotensin II (Ang II). (a) Reverse transcription quantitative polymerase chain reaction and (b and c) Western blot analysis were conducted to detect the expression levels of NLRC3 in vascular smooth muscle cells (VSMCs) after being treated with vector or oe-NLRC3 (n = 3). (d) Cell counting kit-8 assay was used to determine cell viability of VSMCs after being treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3. (n = 3). (e and f) EdU assay was used to assess the proliferation of VSMCs after being treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3 (n = 3). (g and h) Wound healing assay was used to measure the migration of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3 (n = 3). (i and j) Transwell assay tested cell migration capability of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3 (n = 3). Crystal violet was used to stain migration cell. (k and l) Reactive oxygen species production of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3 was measured by 2’,7’-dichlorofluorescin diacetate (n = 3). (m-o) Enzyme-linked immunosorbent assay was employed to determine the levels of malondialdehyde, superoxide dismutase, and glutathione peroxidase in VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, and Ang II (2 µM) + oe-NLRC3 (n = 3). (* P < 0.05, ** P < 0.01, *** P < 0.001. EdU: 5-ethynyl-2’-deoxyuridine, AngII: Angiotensin II, NLRC3: Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3.)
NLRC3 regulates the TRAF6/NF-κB axis to suppress cell growth and migration elicited by Ang II
In a previous study, NLRC3 was found to regulate the NF-κB signaling pathway.[15,23] First, we transfected the overexpression of TRAF6 into VSMCs, which resulted in increased mRNA (P < 0.001) and protein (P < 0.01) levels of TRAF6 [Figure 4a-c]. The overexpression of NLRC3 suppressed cell viability and proliferation in VSMCs treated with Ang II, whereas the overexpression of TRAF6 counteracted the effect of NLRC3 on cell survival (P < 0.01) and proliferation (P < 0.001) [Figure 4d-f]. In addition, the inhibitory effect of NLRC3 on cell migration was attenuated by the overexpression of TRAF6 in VSMCs induced by Ang II (P < 0.01) [Figure 4g-j]. Our findings demonstrated that NLRC3 inhibited cell growth and migration dependent on Ang II by regulating TRAF6.
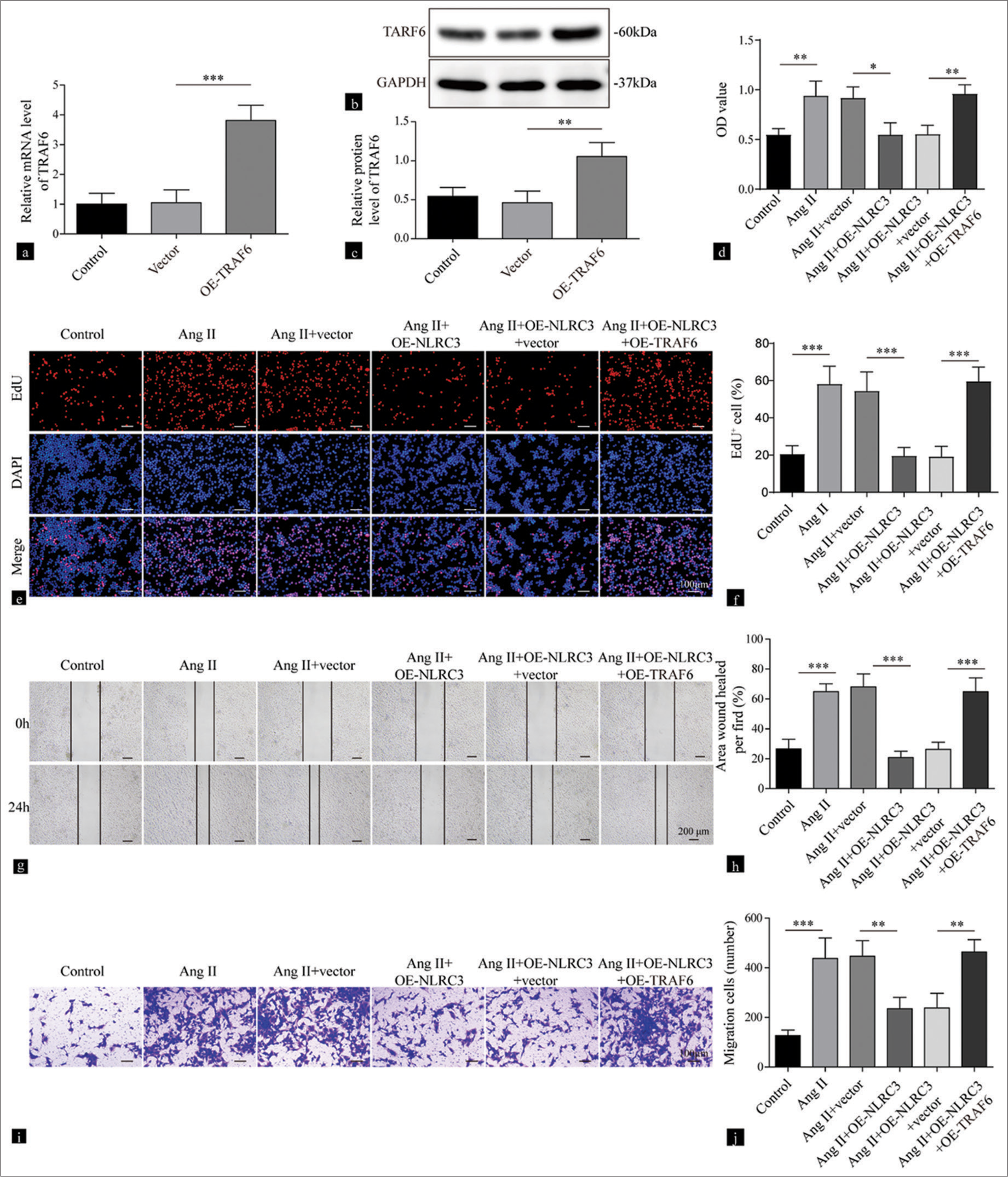
- NLRC3 regulates the TRAF6/nuclear factor-kappa B axis to suppress the proliferation and migration of vascular smooth muscle cells (VSMCs) treated with Angiotensin II (Ang II). (a) Reverse transcription quantitative polymerase chain reaction and (b and c) Western blot analysis were performed to detect the expression levels of TRAF6 in VSMCs treated with vector or oe-TRAF6 (n = 3). (d) Cell viability of VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, Ang II (2 µM) + oe-NLRC3, Ang II (2 µM) + oe-NLRC3+ vector, and Ang II (2 µM) + oeNLRC3 + oe-TRAF6 was evaluated by Cell counting kit-8 assay (n = 3). (e and f) Proliferation of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, Ang II (2 µM) + oe-NLRC3, Ang II (2 µM) + oe-NLRC3+vector, and Ang II (2 µM) + oe-NLRC3+ oe-TRAF6 was assessed by EdU assay (n = 3). (g and h) Migration of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, Ang II (2 µM) + oe-NLRC3, Ang II (2 µM) + oe-NLRC3 + vector, and Ang II (2 µM) + oe-NLRC3+oe-TRAF6 at 0 and 24 h was evaluated by wound-healing assay (n = 3). (i and j) Transwell assay was performed to measure the migration capacity of the VSMCs treated with vehicle, Ang II (2 µM), Ang II (2 µM) + vector, Ang II (2 µM) + oe-NLRC3, Ang II (2 µM) + oe-NLRC3 + vector, and Ang II (2 µM) +oe-NLRC3 + oe-TRAF6 (n = 3) Crystal violet was used to stain migration cell. (* P < 0.05, ** P < 0.01, *** P < 0.001. EdU: 5-ethynyl-2’-deoxyuridine, AngII: Angiotensin II, NLRC3: Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3, TRAF6: tumor necrosis factor receptor-associated factor 6.)
NLRC3 regulates the TRAF6/NF-κB axis to suppress oxidative stress induced by Ang II
We further investigated the roles of the TRAF6/NF-κB axis on oxidative stress mediated by Ang II. The treatment of Ang II promoted the production of ROS in VSMCs and was repressed by the overexpression of NLRC3, and this phenomenon was reversed by the upregulation of TRAF6 (P < 0.001) [Figure 5a and b]. The increase in MDA (P < 0.01), the reduction in SOD (P < 0.01), and the decline in GSH-PX (P < 0.01) elicited by Ang II were reversed by the overexpression of NLRC3, while the overexpression of TRAF6 led to the elevation in MDA (P < 0.05), the reduction in SOD (P < 0.05), and the decrease in GSH-PX (P < 0.05) [Figure 5c-e]. Further analysis of IκBα and p65 phosphorylation demonstrated that Ang II enhanced the phosphorylation of IκBα and p65, which was repressed by NLRC3 (P < 0.001). However, the overexpression of TRAF6 further promoted their phosphorylation (P < 0.01) [Figures 5f-h]. Collectively, these results demonstrated that NLRC3 inhibited oxidative stress induced by Ang II in VSMCs by suppressing the TRAF6/NF-κB axis.
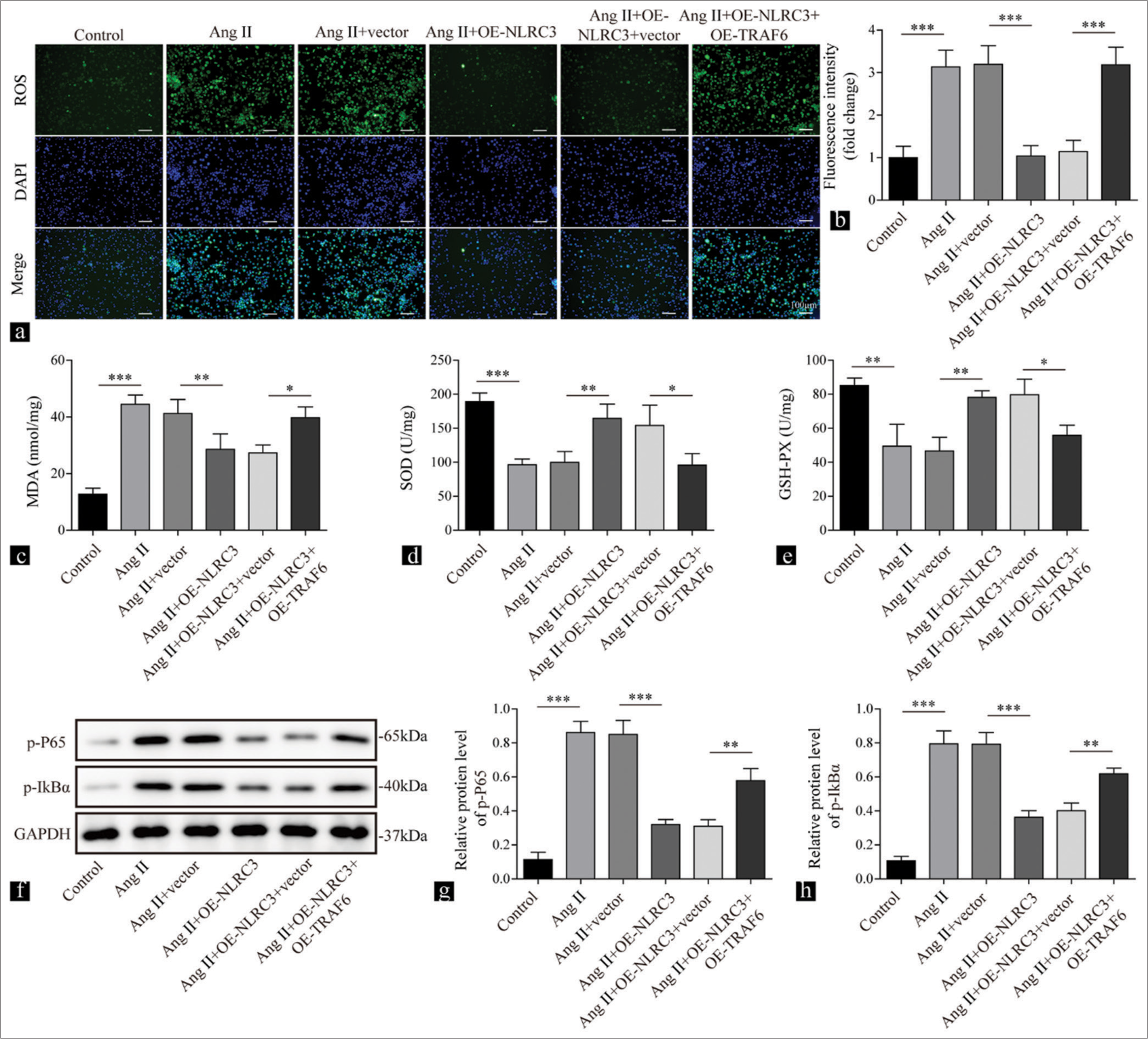
- NLRC3 regulates the TRAF6/nuclear factor-kappa B axis to suppress oxidative stress induced by Angiotensin II (Ang II). Vascular smooth muscle cells were treated with Ang II (2 µM), Ang II (2 µM) + vector, Ang II (2 µM) + oe-NLRC3, Ang II (2 µM) + oe-NLRC3 + vector, and Ang II (2 µM) + oe-NLRC3 + oe-TRAF6. (a and b) Reactive oxygen species production measured by 2’,7’-dichlorofluorescin diacetate in the treated cells (n = 3). (c-e) Results of Enzyme-linked immunosorbent assay showing the levels of malondialdehyde, superoxide dismutase, and Glutathione peroxidase in the treated cells (n = 3). (f-h) Western blot analysis was conducted to determine the levels of phosphorylated p65 and inhibitory subunit of nuclear factor-kappa B alpha in the treated cells (n = 3). (* P < 0.05, ** P < 0.01, *** P < 0.001. EdU: 5-ethynyl-2’-deoxyuridine, AngII: Angiotensin II, NLRC3: Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3, TRAF6: tumor necrosis factor receptor-associated factor 6. )
DISCUSSION
Oxidative stress is deeply intertwined with the pathophysiology of hypertension.[24] However, its mechanisms are not well characterized. Recent studies have identified several potential mechanisms that drive vascular remodeling, including mechanical stimuli and inflammatory signals.[13,25,26] The migration and invasion of VSMCs are also involved in hypertension-related remodeling of vascular system.[27] Studies have also demonstrated the involvement of oxidative stress and lipid oxidation in vascular remodeling.[28,29] Excessive levels of ROS have been found to potentially cause vascular remodeling in various cardiovascular diseases by affecting endothelial dysfunction, inflammation, and VSMCs proliferation or migration.[30,31] The present study sheds light on the potential role of Nur77 in suppressing hypertensive oxidative stress by promoting the transcription of NLRC3, which regulates the TRAF6/NF-κB axis.
Nur77 is known to regulate inflammation and oxidative stress.[10,11,13,32] Pharmacological activation of Nur77 by 6-mercaptopurine repressed inflammation and bone morphogenetic protein signaling, which repressed vascular remodeling.[33] 5-Aminosalicylic acid was shown to mitigate pulmonary arterial hypertension in rats by upregulating the expression of Nur77.[34] In addition, Nur77 inhibited the proliferation of vascular muscle cells.[33,35] Nur77 also suppressed the invasiveness of vascular muscle cells, which is a characteristic response of VSMCs to vascular injury and regeneration.[36,37] In addition, a recent study showed that Nur77 reduced ROS production and alleviate endothelial dysfunction by increasing NO production activating antioxidant pathways in vascular endothelium.[11] Other studies have shown that the knockout of Nur77 could aggravate oxidative stress induced by Ang II in VSMCs.[14,38,39] In the present study, the overexpression of Nur77 repressed growth and oxidative homeostasis mediated by Ang II in VSMCs.
Nur77, as a transcription factor, regulates cellular functions and disease progression by controlling the transcription of its target genes.[10,40] However, the targets of Nur77 transcription involved in hypertensive oxidative stress are unclear. Our current study using JASPAR software prediction displayed NLRC3 as a potential transcriptional target. Further, validation demonstrated that Nur77 initiated the transcription of NLRC3 and increased the expression of NLRC3. Subsequently, we evaluated the function of NLRC3 in oxidative stress induced by Ang II. These results fitted with the previous studies demonstrating the suppressive activity of NLRC3 in attenuating oxidative stress in different hypertension models.[17] The most well-defined downstream pathway that is negatively regulated by NLRC3 is the NF-κB pathway.[16,41,42] In a recent study, the loss of NLRC3 led to the dysregulated activation of the NF-κB/HIF-1α pathway with excessive inflammation, which aggravated pulmonary hypertension.[43] Furthermore, NF-κB signaling contributes to oxidative stress mainly through its immunomodulation and inflammatory regulatory activity.[39,44] Pharmaceutical inhibition of NF-κB has been shown to mitigate pulmonary arterial hypertension.[45] In a previous study, the knockdown of TRAF6 inhibited the proliferation of VSMCs and inflammation.[46] In addition, the inhibition of lncRNA MINCR antagonized lipopolysaccharide-induced acute injury and inflammation through the TRAF6/NF-κB pathways.[47] Another study suggested that NLRC3 repressed NF-κB activation by deubiquitinating TRAF6.[48] Consistently, our current data indicated that NLRC3 upregulation mediated Ang II-mediated VSMCs abnormality, while the activation of NF-κB signaling (TRAF6 overexpression) reversed the suppressive role of NLRC3 on cell proliferation and oxidative stress.
In summary, our results indicated that the overexpression of Nur77 induced the transcriptional upregulation of NLRC3, which inhibited growth and redox imbalance dependent on Ang II in VSMCs by deactivating NF-κB signaling. Our findings suggest a previously undefined Nur77/NLRC3/TRAF6/NF-κB axis that maintains oxidative homeostasis and growth and migration of VSMCs in hypertension. Our study suggests a link between factors that are known to regulate vascular oxidative stress. This understanding provides innovative perspectives into the pathogenesis and progression of hypertension. Our findings may also help identify potential therapeutic targets for hypertension.
SUMMARY
In this study, putative-binding sites for Nur77 on the NLRC3 promoter region were revealed through bioinformatics analysis. Meanwhile, Nur77 was found to upregulate the expression of NLRC3 expression and repress the TRAF6/NF-κB signaling pathway. These phenomena resulted in the reduction in oxidative stress induced by Ang II, which ameliorated hypertension. Our results provide a new insight into mechanisms underlying hypertension induced by Ang II and indicated potential therapeutic targets for the treatment of hypertension.
AVAILABILITY OF DATA AND MATERIALS
Data sharing is not applicable to this article as no datasets were generated or analyzed during the present study.
ABBREVIATIONS
Ang II: Angiotensin II
DAPI: 4’-6-diamidino-2-phenylindol
DCFH-DA: 2’,7’-dichlorofluorescin diacetate
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase
GSH-PX: Glutathione peroxidase
HRP: Horseradish peroxidase
MDA: Malondialdehyde
NF-κB: Nuclear factor-kappa B alpha
NLRC3: Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 3
Nur77: Nuclear receptor subfamily 4 group a member 1
PBS: Phosphate-buffered saline
qPCR: Quantitative polymerase chain reaction
ROS: Reactive oxygen species
SDS–PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis
SOD: Superoxide dismutase
SYBR: Synergetic Binding Reagent
TBST: Tris buffered saline plus tween-20
TRAF6: Tumor necrosis factor receptor-associated factor 6
VSMCs: Vascular smooth muscle cells
AUTHOR CONTRIBUTIONS
LS: Conceptualization, methodology; writing-original draft, funding acquisition; FL: Validation, formal analysis; KX: Resources, data curation; LLZ: Visualization, supervision; DZ: Investigation, project administration; ZQY: Writing - review and editing. All authors have read and approved the final version of the manuscript. All authors have contributed significantly to the work and has agreed to be responsible for every aspect of it.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
No patients or animals involvement, informed consent not required.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This work was supported by Changsha Nature Fund project (No.Kq2007043)
References
- Hypertension in the elderly: What we need to know. Hipertens Riesgo Vasc. 2021;38:91-8.
- [CrossRef] [PubMed] [Google Scholar]
- Hypertension and heart failure: Prevention, targets, and treatment. Cardiol Clin. 2022;40:237-44.
- [CrossRef] [PubMed] [Google Scholar]
- Blood pressure and the brain: The neurology of hypertension. Pract Neurol. 2020;20:100-8.
- [CrossRef] [PubMed] [Google Scholar]
- Oxidative stress and hypertension. Circ Res. 2021;128:993-1020.
- [CrossRef] [PubMed] [Google Scholar]
- Redox stress defines the small artery vasculopathy of hypertension: How do we bridge the bench-to-bedside gap? Circ Res. 2017;120:1721-3.
- [CrossRef] [PubMed] [Google Scholar]
- Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141-8.
- [CrossRef] [PubMed] [Google Scholar]
- The future challenge of Reactive Oxygen Species (ROS) in hypertension: From bench to bed side. Int J Mol Sci. 2017;18:1988.
- [CrossRef] [PubMed] [Google Scholar]
- Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16:431.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear receptor Nur77: Its role in chronic inflammatory diseases. Essays Biochem. 2021;65:927-39.
- [CrossRef] [PubMed] [Google Scholar]
- Nur77 mitigates endothelial dysfunction through activation of both nitric oxide production and anti-oxidant pathways. Redox Biol. 2024;70:103056.
- [CrossRef] [PubMed] [Google Scholar]
- Nur77 downregulation triggers pulmonary artery smooth muscle cell proliferation and migration in mice with hypoxic pulmonary hypertension via the Axin2-beta-catenin signaling pathway. Vascul Pharmacol. 2016;87:230-41.
- [CrossRef] [PubMed] [Google Scholar]
- The orphan nuclear receptor Nur77 inhibits low shear stress-induced carotid artery remodeling in mice. Int J Mol Med. 2015;36:1547-55.
- [CrossRef] [PubMed] [Google Scholar]
- Nur77 attenuates inflammatory responses and oxidative stress by inhibiting phosphorylated IκB-α in Parkinson's disease cell model. Aging (Albany NY). 2020;12:8107-19.
- [CrossRef] [PubMed] [Google Scholar]
- NF-kappaB signaling-mediated activation of WNK-SPAK-NKCC1 cascade in worsened stroke outcomes of Ang II-hypertensive mice. Stroke. 2022;53:1720-34.
- [CrossRef] [PubMed] [Google Scholar]
- Overview of the anti-inflammatory function of the innate immune sensor NLRC3. Mol Immunol. 2023;153:36-41.
- [CrossRef] [PubMed] [Google Scholar]
- NLRC3 inhibits PDGF-induced PASMCs proliferation via PI3KmTOR pathway. J Cell Physiol. 2020;235:9557-67.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic NF-kappaB blockade improves renal angiotensin II type 1 receptor functions and reduces blood pressure in Zucker diabetic rats. Cardiovasc Diabetol. 2015;14:76.
- [CrossRef] [PubMed] [Google Scholar]
- Conditional targeting of tumor necrosis factor receptor-associated factor 6 reveals opposing functions of Toll-like receptor signaling in endothelial and myeloid cells in a mouse model of atherosclerosis. Circulation. 2012;126:1739-51.
- [CrossRef] [PubMed] [Google Scholar]
- miR-634 inhibits human vascular smooth muscle cell proliferation and migration in hypertension through Wnt4/beta-catenin pathway. Front Biosci (Landmark Ed). 2021;26:395-404.
- [CrossRef] [PubMed] [Google Scholar]
- Ultrasmall copper-based nanoparticles for reactive oxygen species scavenging and alleviation of inflammation related diseases. Nat Commun. 2020;11:2788.
- [CrossRef] [PubMed] [Google Scholar]
- NLRC3 expression in macrophage impairs glycolysis and host immune defense by modulating the NF-kappaB-NFAT5 complex during septic immunosuppression. Mol Ther. 2023;31:154-73.
- [CrossRef] [PubMed] [Google Scholar]
- Hypertension and physical exercise: The role of oxidative stress. Medicina (Kaunas). 2016;52:19-27.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of vascular remodeling in hypertension. Am J Hypertens. 2021;34:432-41.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear receptor Nur77 inhibits vascular outward remodelling and reduces macrophage accumulation and matrix metalloproteinase levels. Cardiovasc Res. 2010;87:561-8.
- [CrossRef] [PubMed] [Google Scholar]
- Pathophysiology of vascular remodeling in hypertension. Int J Hypertens. 2013;2013:808353.
- [CrossRef] [Google Scholar]
- Oxidative stress, vascular remodeling, and vascular inflammation in hypertension. Int J Hypertens. 2013;2013:710136.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular oxidative stress: Impact and therapeutic approaches. Front Physiol. 2018;9:1668.
- [CrossRef] [PubMed] [Google Scholar]
- NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol Res. 2016;114:110-20.
- [CrossRef] [PubMed] [Google Scholar]
- Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid Redox Signal. 2014;20:164-82.
- [CrossRef] [PubMed] [Google Scholar]
- Nur77 attenuates inflammasome activation by inhibiting caspase-1 expression in pulmonary vascular endothelial cells. Am J Respir Cell Mol Biol. 2021;65:288-99.
- [CrossRef] [PubMed] [Google Scholar]
- Prevention of progression of pulmonary hypertension by the Nur77 agonist 6-mercaptopurine: Role of BMP signalling. Eur Respir J. 2019;54:1802400.
- [CrossRef] [PubMed] [Google Scholar]
- 5-aminosalicylic acid attenuates monocrotaline-induced pulmonary arterial hypertension in rats by increasing the expression of Nur77. Inflammation. 2017;40:806-17.
- [CrossRef] [PubMed] [Google Scholar]
- Nur77 suppresses pulmonary artery smooth muscle cell proliferation through inhibition of the STAT3/Pim-1/NFAT pathway. Am J Respir Cell Mol Biol. 2014;50:379-88.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular smooth muscle cell motility: From migration to invasion. Exp Clin Cardiol. 2010;15:e75-85.
- [Google Scholar]
- Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler Thromb Vasc Biol. 2018;38:1969-85.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear receptor Nur77 protects against oxidative stress by maintaining mitochondrial homeostasis via regulating mitochondrial fission and mitophagy in smooth muscle cell. J Mol Cell Cardiol. 2022;170:22-33.
- [CrossRef] [PubMed] [Google Scholar]
- TLR2 regulates angiotensin II-induced vascular remodeling and EndMT through NF-kappaB signaling. Aging (Albany NY). 2020;13:2553-74.
- [CrossRef] [PubMed] [Google Scholar]
- Key functions and therapeutic prospects of Nur77 in inflammation related lung diseases. Am J Pathol. 2019;189:482-91.
- [CrossRef] [PubMed] [Google Scholar]
- NLRC3 negatively regulates CD4+ T cells and impacts protective immunity during Mycobacterium tuberculosis infection. PLoS Pathog. 2018;14:e1007266.
- [CrossRef] [PubMed] [Google Scholar]
- NLRC3 alleviates hypoxia/reoxygenation induced inflammation in RAW264.7 cells by inhibiting K63-linked ubiquitination of TRAF6. Hepatobiliary Pancreat Dis Int. 2020;19:455-60.
- [CrossRef] [PubMed] [Google Scholar]
- NLRC3 deficiency promotes hypoxia-induced pulmonary hypertension development via IKK/NF-kappaB p65/HIF-1alpha pathway. Exp Cell Res. 2023;431:113755.
- [CrossRef] [PubMed] [Google Scholar]
- Importance of endothelial NF-kappaB signalling in vascular remodelling and aortic aneurysm formation. Cardiovasc Res. 2013;97:106-14.
- [CrossRef] [PubMed] [Google Scholar]
- Resveratrol inhibits monocrotaline-induced pulmonary arterial remodeling by suppression of SphK1-mediated NF-kappaB activation. Life Sci. 2018;210:140-9.
- [CrossRef] [PubMed] [Google Scholar]
- miR-146-5p restrains calcification of vascular smooth muscle cells by suppressing TRAF6. Open Med (Wars). 2022;17:1515-27.
- [CrossRef] [PubMed] [Google Scholar]
- Depression of lncRNA MINCR antagonizes LPS-evoked acute injury and inflammatory response via miR-146b-5p and the TRAF6-NFkB signaling. Mol Med. 2021;27:124.
- [CrossRef] [PubMed] [Google Scholar]
- The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol. 2012;13:823-31.
- [CrossRef] [PubMed] [Google Scholar]




