


Translate this page into:
Transglutaminase 2 inhibition ameliorates cardiac fibrosis in myocardial infarction by inducing M2 macrophage polarization in vitro and in vivo

*Corresponding author: Qiying Chen, Department of Cardiology, Huashan Hospital, Fudan University, Shanghai, China. chenqiying202202@163.com
-
Received: ,
Accepted: ,
How to cite this article: Maimaitijiang A, Huang Q, Wu Y, Sun S, Chen Q. Transglutaminase 2 inhibition ameliorates cardiac fibrosis in myocardial infarction by inducing M2 macrophage polarization in vitro and in vivo. CytoJournal. 2024;21:58. doi: 10.25259/Cytojournal_32_2024
Abstract
Objective:
Macrophages perform vital functions in cardiac remodeling after myocardial infarction (MI). Transglutaminase 2 (TG2) participates in fibrosis. Nevertheless, the role of TG2 in MI and mechanisms underlying macrophage polarization are unclear. This study aimed to discover the functions and possible mechanisms of TG2 in MI.
Material and Methods:
C57BL/6 mice were classified into three groups (six mice per group): Sham, MI, and MI+GK921 groups. GK921 acts as a TG2 inhibitor. Cardiac function, myocardial cell apoptosis, fibrosis, and macrophage phenotype in mouse experiments were detected through echocardiography, terminal deoxynucleotidyl transferase dUTP nick end labeling, Masson staining, immunofluorescence, and flow cytometry, respectively. The in vitro study involved the treatment of mouse cardiac fibroblasts isolated from mice with transforming growth factor β1 (TGF-β1) and evaluation of fibrosis through the detection of the expressions of fibrosis-associated proteins using Western blot. Bone marrow-derived macrophages (BMDMs) obtained from mice were triggered by interleukin (IL)-4, and the type of macrophages was determined through flow cytometry.
Results:
In in vivo experiments, GK921 substantially improved cardiac injury and fibrosis, induced M2 macrophage polarization, and suppressed the TGF-β1/small mother against decapentaplegic 3 (Smad3) pathway in MI mice. Moreover, TG2 knockdown considerably decreased the expressions of fibrosis-associated proteins in TGF-β1-triggered mouse cardiac fibroblasts, which indicates the repressive effect of TG2 knockdown on fibrosis. In addition, the inhibition effect of TG2 downregulation on the TGF-β1/Smad3 pathway was proven in TGF-β1-treated mouse cardiac fibroblasts in vitro. Moreover, TG2 inhibition remarkably increased M2 macrophage polarization in IL-4-induced BMDMs.
Conclusion:
TG2 inhibition facilitated M2 macrophage polarization to provide protection against MI-caused cardiac fibrosis in mice, and these effects may be attained through modulation of the TGF-β1/Smad3 pathway.
Keywords
Myocardial infarction
Transglutaminase 2
Macrophage polarization
Transforming growth factor β1/small mother against decapentaplegic 3 pathway

INTRODUCTION
The cardiomyocyte loss and collagen deposition caused by myocardial infarction (MI) lead to left ventricular remodeling and serve as functional and structural bases for the initiation and development of heart failure.[1,2] Different from other organs, the heart damage caused by MI cannot be repaired through cardiac regeneration of muscle cells.[3] Instead, the necrotic myocardium is replaced by fibrous scar.[4] Cardiac fibrosis is a common characteristic observed after MI involving fibrosis processes, such as fibroblast proliferation, fibroblast differentiation into myofibroblasts, extracellular matrix deposition, and cardiac remodeling.[5,6] MI remains a dominating cause of mortality among heart diseases worldwide.[7-9] Therefore, more therapeutic agents for MI patients should be discovered.
Transglutaminase 2 (TG2) is one of the Ca2+-dependent enzymes, and it exerts multifunctions, including protein disulfide isomerization, transamidation, and protein kinase activation.[10,11] TG2 is commonly expressed in a variety of tissues and plays a key role in various diseases, such as autoimmune and neurodegenerative diseases.[12] Notably, TG2 contributes to the pathological mechanisms of tissue fibrosis, including the lungs and kidneys.[13,14] The inhibition of the activity of TG2 can attenuate cardiac fibrosis in the acute MI model.[15] Moreover, TG2 is implicated in the protection of cardiomyocytes against ischemia/reperfusion-caused injury.[16] Nevertheless, the role of TG2 in MI-induced cardiac fibrosis still needs exploration.
Drastic tissue inflammation initiates cardiac repair after MI, and multiple molecular and cellular factors influence post-MI wound repair.[17,18] Tissue regeneration shows a close relation to macrophage phenotype or polarization. The two main phenotypes of macrophages include classical M1 (proinflammatory) and M2 macrophages (anti-inflammatory).[19] M2 macrophages can release large amounts of transforming growth factor β (TGF-β), interleukin (IL)-10, and vascular endothelial growth factor, which accelerate anti-inflammation response and repair.[20,21] Moreover, M2 macrophage polarization has been implicated in fibrosis after acute heart transplant.[22] In addition, M2 macrophages are associated with liver fibrosis.[23] TGF-β1 serves as a master switch for inducing fibrosis, regulating extracellular matrix-correlated gene expression, and activating the small mother against decapentaplegic 2/3 (Smad2/3) and TGF-β receptors to stimulate cardiac fibroblasts;[24] the TGF-β1/Smad3 pathway has been proven as a common pathway in tissue fibrosis development.[25] Therefore, we investigated the roles of M2 macrophages and the TGF-β1/Smad3 pathway post-MI in mice.
The present research aimed to explore the functional role of TG2 in post-MI fibrosis and its underlying mechanisms, with a focus on M2 macrophage polarization and the TGF-β1/Smad3 pathway.
MATERIAL AND METHODS
Study design
For in vivo experiments, C57BL/6 mice (male, 8–10 weeks) were purchased from the Beijing Vital River Laboratory Animal Technology (Beijing, China), and three groups were set: (1) Sham, (2) MI, and (3) MI+GK921 groups. Two hours before MI induction, mice in the MI+GK921 group were intraperitoneally injected with TG2 inhibitor GK921 (2 mg/Kg; HY-1233, MedChemExpress, Monmouth Junction, NJ, USA). Normal saline was injected into the MI and Sham groups through intraperitoneal injection. The mice were kept for 7 days before undergoing echocardiography and histological studies. On day 8, mice were performed to detect cardiac function. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), Masson’s trichrome staining, and immunofluorescence analysis were conducted for the evaluation of cardiomyocyte apoptosis, cardiac fibrosis, and macrophage polarization in heart tissues, respectively. This study received approval from the ethics committee of Huashan Hospital, Fudan University (No: 2020-JS-574), and all animal procedures complied with approved protocols.
The in vitro experiments involved the stimulation of cardiac fibroblasts with 100 µg/mL TGF-β1 (7754-BH, R and D Systems, Minneapolis, MN, USA) for 24 h to construct a myocardial fibrosis model. Next, the effects of knocking down TG2 on fibrosis were assessed using a myocardial fibrosis cell model. IL-4 (10 ng/mL, 404-ML; R and D Systems, Minneapolis, MN, USA) was used to interfere with bone marrow-derived macrophages (BMDMs) for 24 h to induce macrophage polarization. The evaluation subsequently focused on the effects of TG2 on macrophage polarization.
Animals and MI model
The C57BL/6 mice (n = 20) were housed in a controlled environment with 12/12 h light/dark cycle (40% ± 5% humidity and 20 ± 2°C temperature) and given standard mouse food and water. Two mice were sacrificed through cervical dislocation for the isolation of BMDMs and myocardial fibroblasts, and the remaining mice were used for animal experiments. Specifically, after 7 days of acclimation, the mice were divided into three groups: Sham (n = 6), MI (n = 6), and MI+GK921 groups (n = 6). All mice were anesthetized using 3% isoflurane (R510-22-10, Ruiwode, Shenzhen, China), and artificial respiration was maintained using a positive-pressure respirator (ALC-V8, Alcott Biotech Co., LTD, Shanghai, China) with a frequency of 110 strokes/min and a tidal volume of 20 mL/kg. The heart was accessed through a left thoracotomy, and visualization of the left anterior descending (LAD) artery was achieved using a surgical microscope (YAN-6A model, YuYan, Shanghai, China). At 2 mm below the tip of the left atrial appendage, the LAD was ligated with a 7-0 silk suture. After 30 min of ischemia, the ligation was clipped to achieve reperfusion. Identical surgical procedures were performed for all groups except for the Sham group except, which did not undergo LAD ligation. At 2 h before MI surgery, mice in the MI+GK921 group were intraperitoneally injected with 2 mg/kg GK921. In the MI and Sham groups, the mice received normal saline through intraperitoneal injection before MI surgery.
Echocardiography
Seven days after MI surgery, the mice (n = 6 per group) were anesthetized for the echocardiography scans. Cardiac function was assessed using an echocardiography machine (Vevo3100, FUJIFILM, Toronto, Canada). The following parameters were acquired: Left ventricular ejection fraction (LVEF), left ventricular fraction shortening (LVFS), left ventricular volume at end diastole (LV vol: s), and left ventricular internal dimension in systole (LVIDs).
Histological preparation
Seven days after MI surgery, the mouse heart was exposed after the mice received anesthesia. Three mice from each group were selected for TUNEL staining, immunofluorescence, and Masson’s trichrome staining. The hearts were arrested through ventricular injection of 1 mol/L potassium chloride (KCl, P9921, Solarbio, Beijing, China), followed by perfusion using 4% paraformaldehyde (P1110, Solarbio, Beijing, China). Subsequently, the hearts were collected and fixed using 4% paraformaldehyde. In addition, three mice from each group were used for flow cytometry and Western blot. Their hearts were flushed with phosphate buffered saline, frozen in liquid nitrogen, and finally stored at −80°C.
TUNEL staining
Seven days after operation, the extracted hearts were fixed in 4% paraformaldehyde, paraffin embedded, and sliced into 3 µm section. Afterward, the slices were permeabilized with 0.3% Triton X-100 (T8200, Solarbio, Beijing, China) for 20 min and then blocked using 5% bovine serum albumin (BSA, SW3015, Solarbio, Beijing, China) for 30 min at room temperature. Next, the slices were stained using a TUNEL fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (C1088, Beyotime, Shanghai, China), in accordance with the manufacturer’s instructions. Nuclei were stained with 1 µg/mL 4,6-diamino-2-phenyl indole (DAPI, ab285390, Abcam, Cambridge, MA, USA). Finally, a fluorescence microscope (DM4B, Leica, Wetzlar, Germany) was used to capture images. The rate of TUNEL-positive cells was quantified using Image J 1.8.0 software (Media Cybernetics, Silver Spring, MD, USA).
Masson’s trichrome staining and immunofluorescence analysis
Masson’s trichrome staining involved staining 3 µm paraffin-embedded myocardial tissue samples from infarction area using Masson’s trichrome kit (C0189S, Beyotime, Shanghai, China) to analyze histological changes. Immunofluorescence assay included the overnight incubation of the sections with M2 macrophage polarization markers, including anti-macrophage mannose receptor (anti-CD206; 1:100; 18704-1-AP, ProteinTech, Rocky Hill, NJ, USA) and anti-arginase-1 (Arg-1); 1:100; 16001-1-AP, ProteinTech, Rocky Hill, NJ, USA) and culturing with the secondary antibody (SA00009-2, ProteinTech, Rocky Hill, NJ, USA) for 1.5 h. After washing, the nuclei in the sections were identified through application of DAPI. Finally, the images of CD206-positive and Arg-1-positive cells were photographed using a microscope (DM4B, Leica, Wetzlar, Germany). Image J 1.8.0 software (Media Cybernetics, Silver Spring, MD, USA) was adopted to analyze the positive-staining cells.
Cells isolation and treatment
Cardiac fibroblasts were isolated from the C57BL/6 mice as previously described.[26] Briefly, dissection was performed to remove the atria and vessels from the heart tissues extracted from the C57BL/6 mice. Then, the hearts were digested and finely fragmented. After centrifugation (400 g, 5 min, 4°C), the cells were suspended and plated. After 1 h, adherent cardiac fibroblasts were expanded through dispersion involving ethylenediaminetetraacetic acid/trypsin solution (T1300, Solarbio, Beijing, China). The isolated mouse cardiac fibroblasts were identified after anti-vimentin staining. Given that the passage number can affect the fibroblast phenotypes, only 1–3 passage fibroblasts were used in the experiments. Myocardial fibrosis was evaluated through incubation of 1–3 passage cardiac fibroblasts with 100 µg/mL TGF-β1 for 24 h.
BMDMs were isolated from the bone marrow of C57BL/6 mice.[27] Briefly, the tibias and femurs were obtained from the mice, and the bone marrow was harvested and filtered through a 70 µ m strainer. The cell pellet was collected after centrifugation for 5 min at 1000 rpm at 4°C. Cells were treated with 20 ng/ml macrophage colony-stimulating factor (400-28, PeproTech, Rocky Hill, NJ, USA) for 3 days. After 5–7 days of differentiation, the BMDMs, which were identified by epidermal growth factor-like module-containing mucin-like hormone receptor-like 1 (F4/80) staining, were used for this experiment. To induce macrophage polarization, we triggered BMDMs with IL-4 (10 ng/mL) for 24 h. All cells were subjected to mycoplasma testing before conducting the experiment to ensure the absence of contamination.
Cell transfection
Small interfering RNA against TG2 (si-TG2#1: 5’-CCACCCACCATATTGTTTGAT-3’, si-TG2#2:5’-CAG TTCGAGGATGGAATCCTGGATA-3’) and its small interfering negative control (si-NC) were designed by GenePharma (Shanghai, China). Cardiac fibroblasts and BMDMs were transfected with 0.5 µg si-NC or si-TG2 using 0.6 µL Lipofectamine 2000 (11668027, Invitrogen, Carlsbad, CA, USA) for 48 h.
Flow cytometry
Macrophage polarization in mice after GK921 and MI treatment was evaluated through flow cytometry after heart tissue digestion. In brief, fresh heart tissues were dissected after rinsing the residual blood. The samples were digested using a mixture of collagenase IV, hyaluronidase, and deoxyribonuclease for 30 min at 37°C. Then, the digested samples were filtrated using a 70 µm nylon mesh and then centrifuged at 600 g for 5 min at 4°C. Next, cell suspensions were stained with FITC-conjugated anti-CD206 (141703, Biolegend, San Diego, CA, USA), and phycoerythrinconjugated anti-mouse F4/80 (111603, Biolegend, San Diego, CA, USA) antibodies. The used antibodies were purchased from Biolegend (San Diego, CA, USA). The M2 macrophages, characterized as CD206+ F4/80+ cells, were identified using a FACSCalibur instrument (BD Biosciences, San Jose, CA, USA). Live cells were gated on F4/80+/CD206+.
Western blot
Total proteins were isolated from the heart tissues, mouse cardiac fibroblasts, and BMDMs. The proteins were separated through 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis by electrophoresis, and then, protein transfer was conducted onto the polyvinylidene fluoride membranes (1620264, Bio-Rad, Hercules, CA, USA). The membranes were blocked using 5% BSA (2 h, room temperature), and specific primary antibodies were applied for incubation overnight at 4°C, including anti-CD206 (Cat#24595; 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-α-smooth muscle actin (anti-α-SMA, E-AB-34268; 1:500; Elabscience, Wuhan, China), anti-Arg-1 (Cat#93668; 1:1000; Cell Signaling Technology), anti-collagen I (ab138492; 1:1000; Abcam), anti-IL-10 (ab133575; 1:1000; Abcam), anti-TGF-β1 (sc-130348; 1:200; Santa Cruz Biotechnology), anti-phospho-Smad3 (p-Smad3, ab52903; 1:2000; Abcam), anti-collagen III (ab184993; 1:1000; Abcam), anti-Smad3 (ab40854; 1:1000; Abcam), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab9485; 1:1000; Abcam), followed by the treatment with goat anti-rabbit secondary antibody (ab150077; 1:10000; Abcam). The protein bands were visualized using an enhanced chemiluminescence kit (Millipore, Burlington, MA, USA) in a Fluorchem HD2 imaging system (BioTechne, Minneapolis, MN, USA). GAPDH was used as the internal reference, and the gray scale values of target proteins were analyzed using Image J (Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
The data were denoted as the mean ± standard deviation and analyzed using GraphPad Prism 8.0 software (Graphpad, San Diego, CA, USA). One-way analysis of variance followed with a post hoc Tukey’s test was performed to determine multiple comparisons, and unpaired t-test was conducted to compare the differences between two groups. P < 0.05 was considered significant.
RESULTS
TG2 inhibition improved MI-induced cardiac dysfunction and myocardial damage
First, we explored the role of TG2 in mice after MI. Post-MI, LVEF, and LVFS exhibited dramatical decreases (P < 0.001), and LV vol: s and LVIDs showed marked increases (P < 0.001) [Figure 1a-e], which indicates that MI caused cardiac dysfunction. However, the treatment with TG2 inhibitor GK921 exhibited a cardioprotective effect and attenuated cardiac dysfunction, as evidenced by the elevated levels of LVEF and LVFS and the diminished LV vol: s and LVIDs (P < 0.05) [Figure 1a-e]. The TUNEL results show the increase of TUNEL positive rate in MI group compared with sham group (P < 0.001) [Figure 1f-g], indicating the facilitation of cardiomyocyte apoptotic after MI. While TG2 inhibition remarkably repressed TUNEL positive rate in MI mice (P < 0.05) [Figure 1f-g], suggesting suppression of cardiomyocyte apoptosis affected by TG2 inhibition. Furthermore, Masson’s trichrome staining revealed that cardiac fibrosis was evidently augmented in mice after MI (P < 0.001, [Figure 1h and i]) and was significantly restrained after GK921 treatment (P < 0.05) [Figure 1h and i].
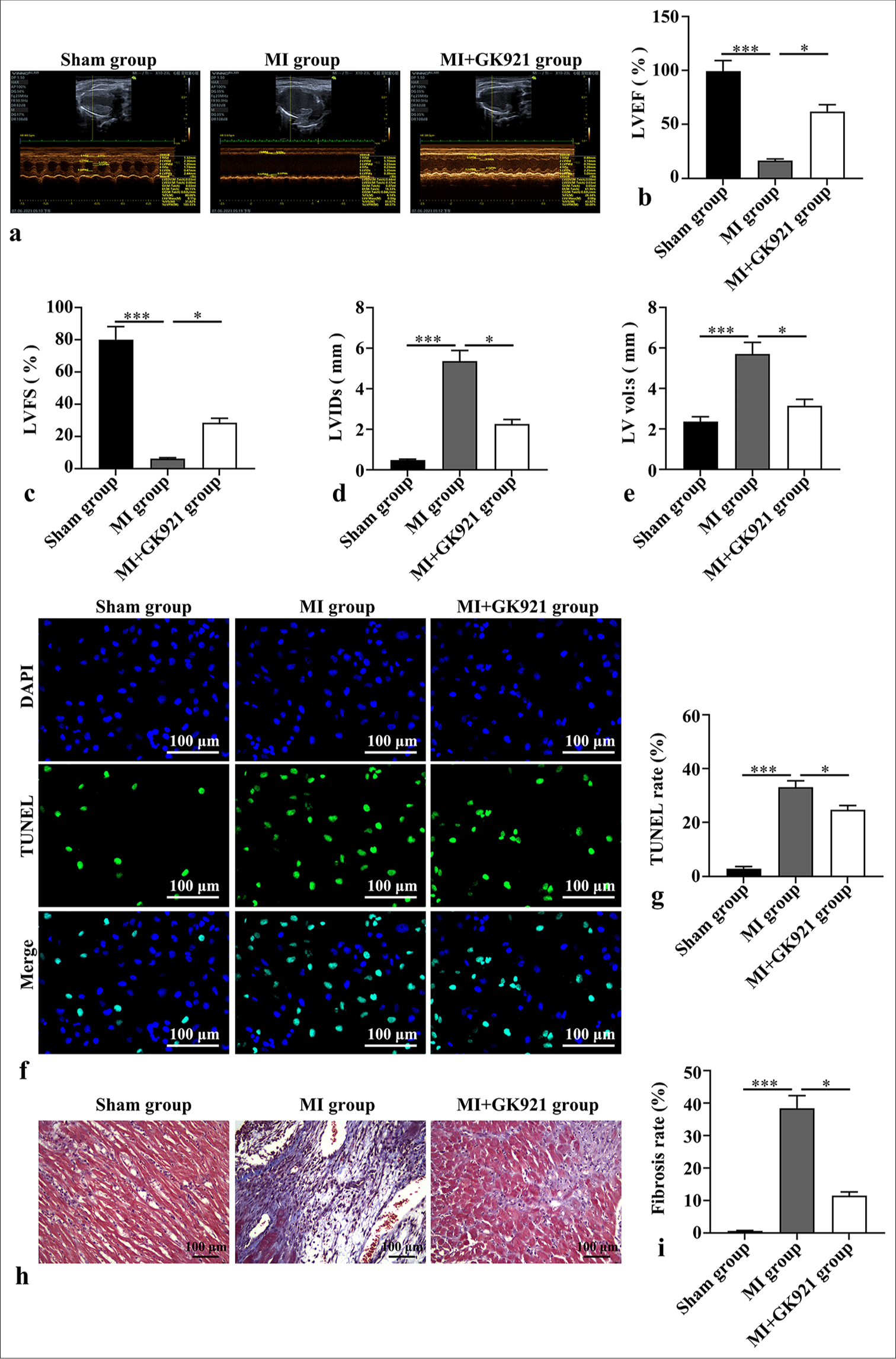
- Transglutaminase 2 inhibition improved myocardial infarction (MI)-induced cardiac dysfunction and myocardial damage. Myocardial infarction model was formed using C57BL/6 mice, and GK921 (a transglutaminase 2 inhibitor) was applied in mouse pre-treatment before myocardial infarction. (a-e) Echocardiography was performed, and cardiac function parameters, including left ventricular ejection fraction (LVEF), left ventricular fraction shortening (LVFS), left ventricular volume at end diastole (LV vol: s), and Left ventricular internal dimension in systole (LVIDs), were detected (n = 6). (f-g) Cardiomyocyte apoptosis was determined through Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (n = 3). Scale bar, 100 µm. TUNEL staining shows green fluorescence, the cell nucleus are stained with 4’,6-diamidino-2-phenylindole (DAPI) and show blue fluorescence. (h-i) Masson’s trichrome staining was used to evaluate cardiac fibrosis in mice (n = 3). (Scale bar, 100 µm. *P < 0.05, and ***P < 0.001. DAPI: 4’,6-diamidino-2-phenylindole.)
TG2 inhibition promoted M2 macrophage polarization in mice after MI
Flow cytometry was performed to analyze the single-cell suspensions obtained from the heart tissues of the three groups to evaluate macrophage differentiation. F4/80+CD206+ macrophages (M2 macrophages) showed a marked enhancement after MI (P < 0.01), and GK921 further increased the percentage of M2 macrophages (P < 0.01) [Figure 2a and b]. In addition, we detected the CD206 and Arg-1 expressions in the heart tissues of the three groups through immunofluorescence staining and Western blot. The results of immunofluorescence revealed the increased expressions of CD206- and Arg-1-positive cells in the GK921+ MI group compared with the MI group (P < 0.01) [Figure 2c-e]. Similarly, the protein levels of CD206 and Arg-1 were dramatically augmented in the MI group compared with the Sham group (P < 0.01), and GK921 further increased CD206 and Arg-1 expressions in MI mice (P < 0.05) [Figure 2f and g].
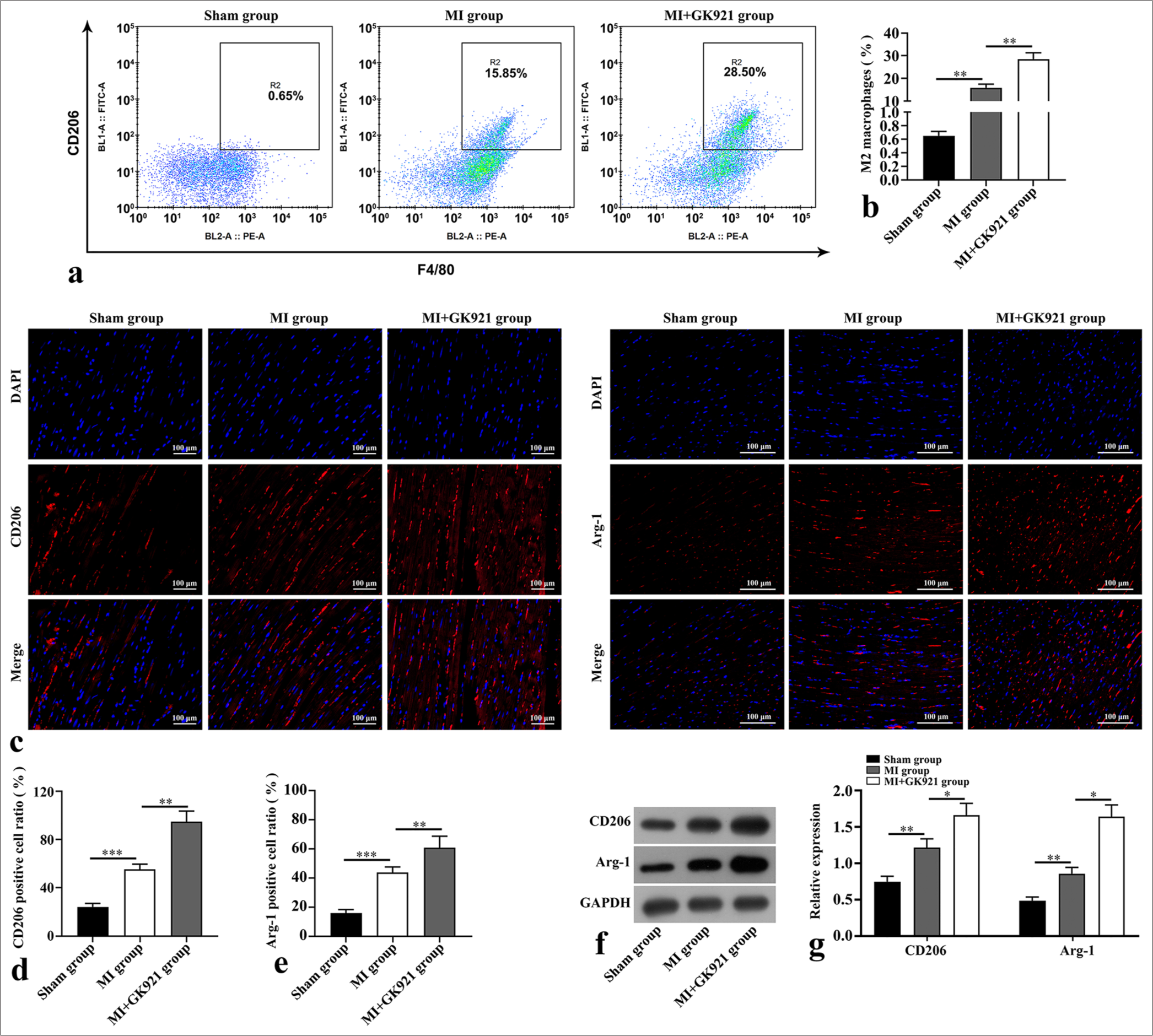
- Transglutaminase 2 inhibition promoted M2 macrophage polarization in mice after myocardial infarction (MI). (a and b) Flow cytometry was performed to assess myocardial epidermal growth factor-like module-containing mucin-like hormone receptor-like 1 (F4/80)+ macrophage mannose receptor (CD206)+ macrophages (n = 3). (c-e) Immunofluorescence staining of M2 macrophage polarization markers in mouse heart tissues (n = 3). Scale bar, 100 µm. The CD206 and arginase-1 (Arg-1) staining exhibits red fluorescence, and 4’,6-diamidino-2-phenylindole (DAPI) staining for labeling cell nuclei shows blue fluorescence. (f and g) Protein expressions of CD206 and Arg-1 in mouse heart tissues were detected using Western blot (n = 3). (*P < 0.05, **P < 0.01, and ***P < 0.001. DAPI: 4’,6-diamidino-2-phenylindole, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.)
TG2 inhibition suppressed the TGF-β1/Smad3 pathway in MI mice
We detected the effects of GK921 on the TGF-β1/Smad3-related protein expression. Our data reveal the significantly augmented TG2 protein expression in the MI group. However, such effect was reversed by GK921 (P < 0.05) [Figure 3a and b]. Western blot results disclose the overtly boosted TGF-β1 level and p-Smad3/Smad3 in mice after MI (P < 0.05), but they were markedly depressed by TG2 inhibition (P < 0.05) [Figure 3c and d]. Thus, TG2 inhibition repressed the activation of TGF-β1/Smad3 pathway induced by MI.
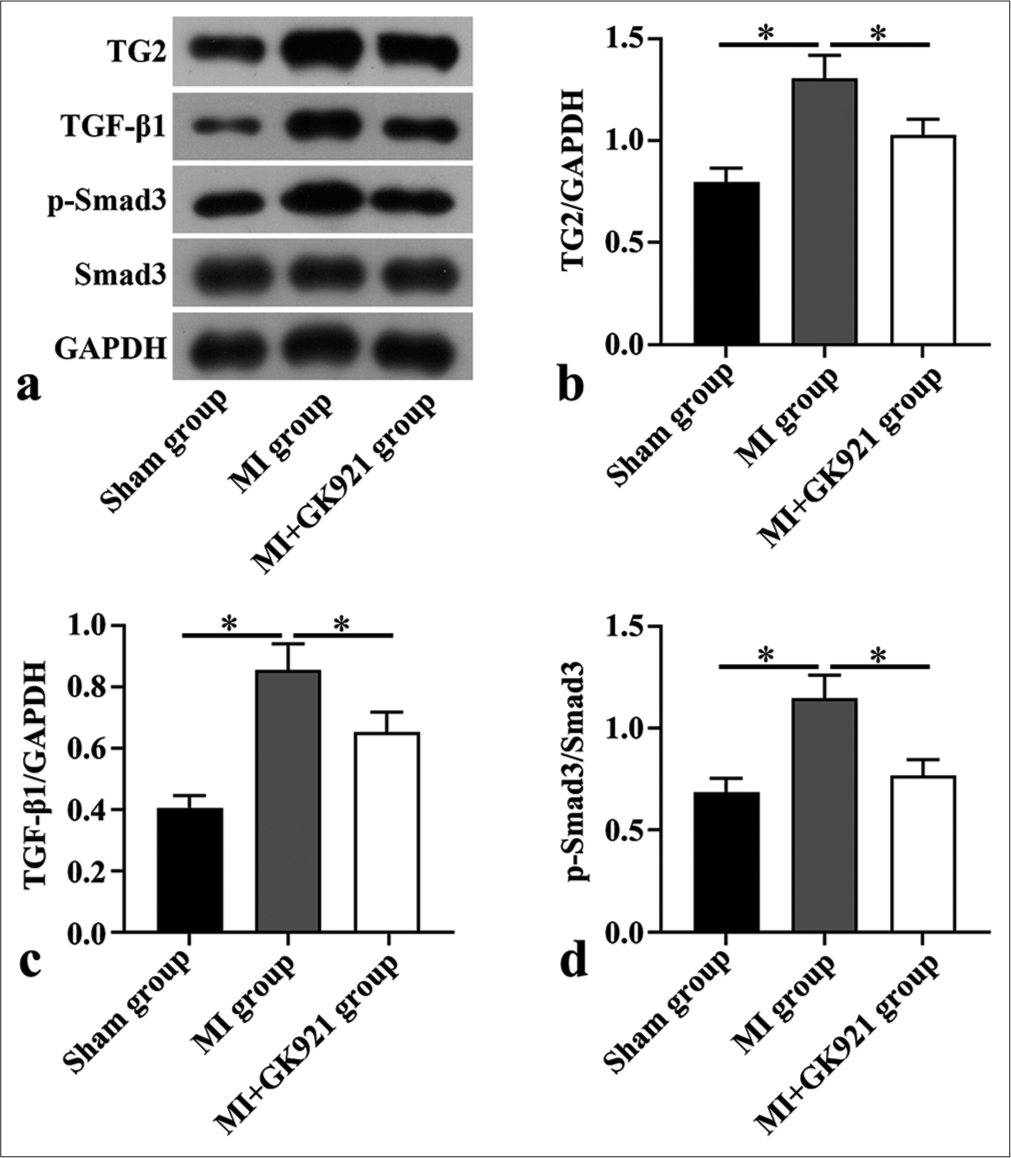
- Transglutaminase 2 inhibition suppressed the transforming growth factor (TGF)-β1/Small mother against decapentaplegic 3 (Smad3) pathway in myocardial infarction (MI) mice. (a) Transglutaminase 2 (TG2), TGF-β1, phosphoSmad3 (p-Smad3), and Smad3 protein expression measured in mouse heart tissues via Western blot (n = 3). (b - d) Quantitative analysis of TG2, TGF-β1, p-Smad3/Smad3 (n = 3). (*P < 0.05. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.)
TG2 inhibition decreased TGF-β1-induced myocardial fibrosis and repressed the TGF-β1/Smad3 pathway in mouse cardiac fibroblasts
Cardiac fibroblasts were isolated from mice and knocked down the TG2 expression using si-TG2. The data show that si-TG2#1 significantly decreased TG2 protein expression compared with si-NC (P < 0.05). However, si-TG2 # 2 did not exhibit this effect [Figure 4a and b]. Thus, we selected si-TG2#1 for the next experiments. In addition, cardiac fibroblasts were treated with TGF-β1 to evaluate myocardial fibrosis. Western blot findings verified that TGF-β1 treatment dramatically boosted the levels of fibrosis-promoting proteins (α-SMA and collagens III and I) (P < 0.05), and knockdown of TG2 reduced the expressions of these proteins in cardiac fibroblasts (P < 0.05) [Figure 4c-f]. Moreover, we found that the increased levels of TGF-β1 and p-Smad3/Smad3 triggered by TGF-β1 treatment were markedly attenuated after downregulating TG2 in cardiac fibroblasts (P < 0.05) [Figure 4g-i].
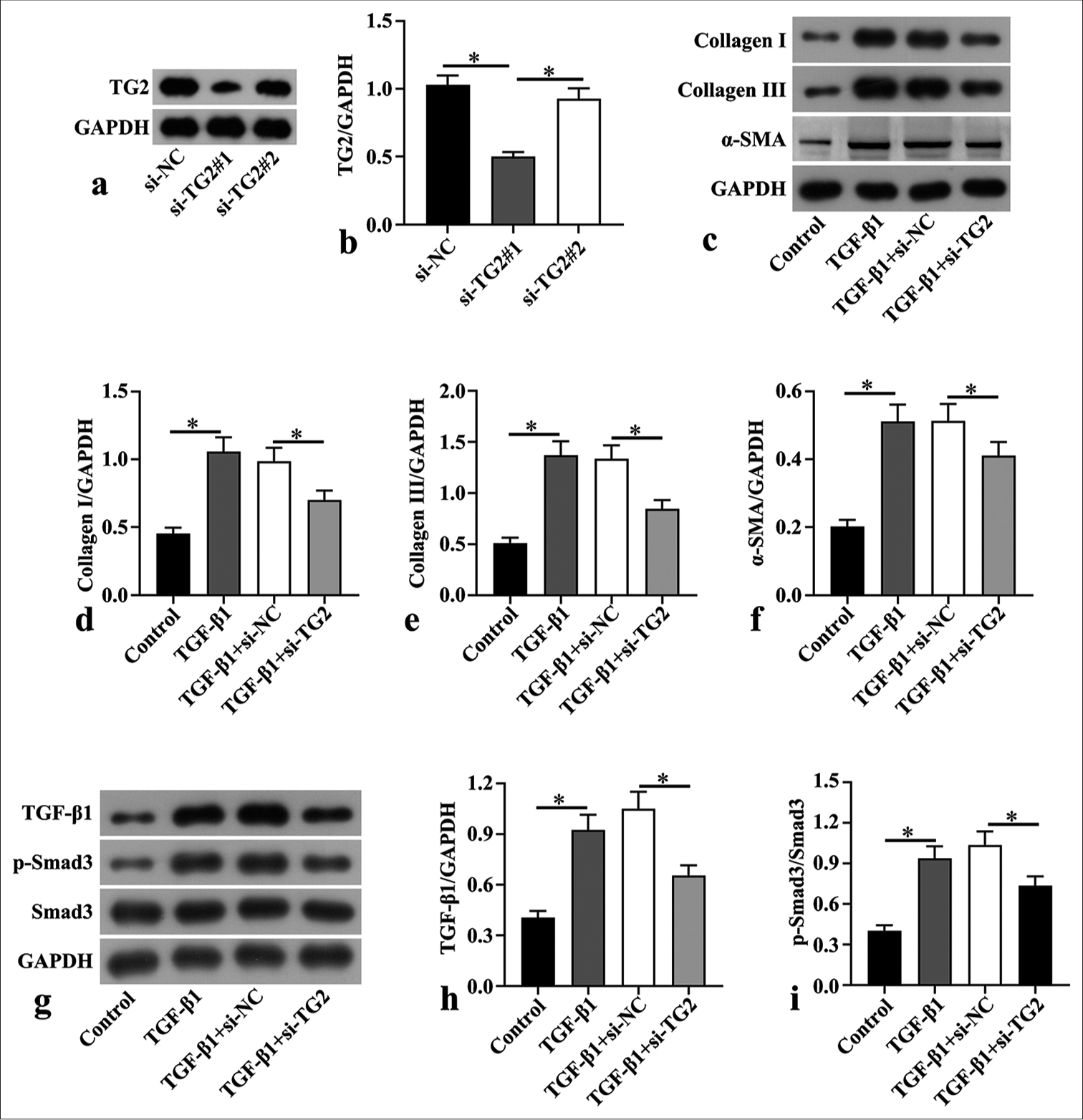
- Transglutaminase 2 downregulation transforming growth factor (TGF)-β1-induced myocardial fibrosis and repressed TGF-β1/small mother against decapentaplegic 3 (Smad3) pathway in mouse cardiac fibroblasts. (a and b) Small interfering negative control (si-NC) and siTransglutaminase 2 (si-TG2) were transfected to mouse cardiac fibroblasts isolated from C57BL/6 mice, and the protein expression of TG2 was detected through Western blot. (c-f) Mouse cardiac fibroblasts were treated with 100 µg/mL TGF-β1 to induce fibrosis, followed by transfection with siNC and si-TG2 for 48 h. Western blot was conducted to determine the levels of fibrosis-associated proteins, including collagen I, collagen III, and α-smooth muscle actin (α-SMA), in mouse cardiac fibroblasts after TGF-β1 treatment and transfection (n = 3). (g-i) Western blot was also used in the assessment of the levels of TGF-β1, phospho-Smad3 (p-Smad3), and Smad3 in mouse cardiac fibroblasts after TGF-β1 treatment and transfection (n = 3). (*P < 0.05. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.)
TG2 inhibition increased M2 macrophage polarization in IL-4-induced BMDMs
According to Western blot results, the protein expressions of M2 makers, including CD206, Arg-1, and IL-10, were markedly elevated in IL-4-triggered BMDMs, which were further enhanced by TG2 knockdown (P < 0.05) [Figure 5a-d].
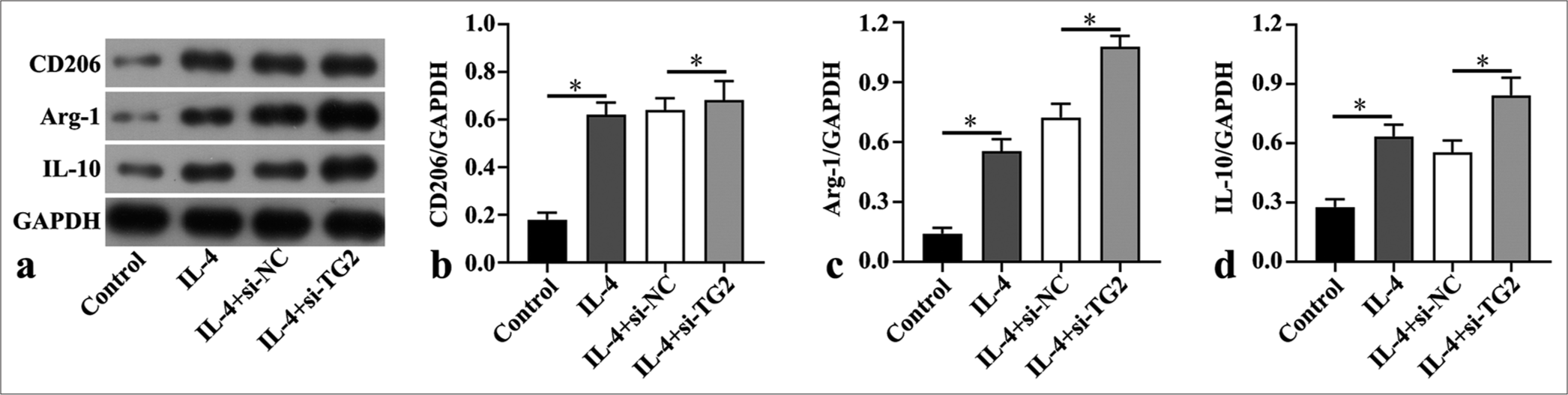
- Transglutaminase 2 inhibition increased M2 macrophage polarization in interleukin (IL)-4-induced bone marrow derived macrophages. (a-d) Western blot was utilized to assess the levels of macrophage mannose receptor (CD206), arginase-1 (Arg-1) and interleukin-10 (IL-10) in interleukin (IL)-4-induced bone marrow derived macrophages after transfection of small interfering negative control (si-NC) and si-Transglutaminase 2 (si-TG2) (n = 3). (*P < 0.05. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.)
DISCUSSION
MI is a life-threatening cardiovascular disease with a high mortality rate worldwide.[28] The effects of TG2 on liver and lung fibrosis have been proven, but its role in cardiac fibrosis remains unclear. More importantly, macrophage polarization is a key regulator in cardiac remodeling post-MI.[29] Previous extensive investigations have further highlighted the participation of the TGF-β1/Smad3 pathway in macrophage activation and fibrosis.[30-32] We used this evidence to explore the function of TG2 in cardiac fibrosis and further investigate its association with macrophage polarization and the TGF-β1/Smad3 pathway post-MI.
TG2 shows a linkage to extracellular matrix remodeling, tissue stability, cell adhesion, apoptosis, and wound healing.[33,34] Moreover, TG2 was discovered in fibrotic kidney tissues from patients or animals,[35-38] and its overexpression resulted in interstitial cardiac fibrosis in mice.[39] Accordingly, in the present study, the MI mouse model was established, and MI mice were pre-treated with GK921. Then, cardiac function, apoptosis, and fibrosis in mice were assessed post-MI. Our results indicate that TG2 inhibition alleviated cardiac dysfunction caused by MI. Furthermore, GK921 treatment reduced the MI-induced myocardial apoptosis and fibrosis, which suggests that TG2 inhibition improved the cardiac dysfunction and fibrosis caused by MI in mice in vivo. A previous study reported that GK921 attenuated bleomycin-induced pulmonary fibrosis in mice but caused no change in the TG2 expression in mouse alveolar epithelial cells MLE-21,[40] which is different from our results. Such variation may be a result of the differences in samples and models. In addition, mouse cardiac fibroblasts were transfected with si-TG2 to explore the role of TG2 in fibrosis in vitro. TG2 down-regulation caused the reversal of the promoting effects of TGF-β1 on fibrosis, consistent with the findings of our in vivo experiments.
Inflammation is a vital process post-MI and closely related to fibrosis.[41] Macrophages played critical roles in wound healing impairment and cardiac remodeling after MI.[29,42] Macrophages contribute to inflammation response and fibrosis.[43] Post-MI mice showed a significant increase in the percentage of anti-inflammatory M2 macrophages following GK921 treatment, which indicates that TG2 inhibition promoted M2 macrophage polarization. si-TG2 was transfected into BMDMs, and the effects of TG2 on the percentage of pro-inflammatory macrophages were explored. The results demonstrated that the pro-inflammatory macrophages were dramatically repressed by downregulating TG2 in BMDMs. These data reveal that TG2 inhibition exerted protective effects on cardiac fibrosis through suppression of the pro-inflammatory macrophages.
TGF-β1 is considered a key regulator in cardiac fibroblast induction through modulation of the TGF-β/Smad pathway.[44-46] Hyperactivation of TGF-β1 can enhance cardiac fibroblast transformation into myofibroblasts and eventually cause cardiomyocytes death, interstitial fibrosis, and augmentation of cardiac stiffness.[47,48] In this study, TGF-β1 showed a high expression in heart tissues from MI mice, and it was attenuated by GK921. GK921 also repressed the level of p-Smad3/Smad3 increased by MI. Similarly, the TGF-β1 levels and p-Smad3/Smad3 ratio were both up-regulated by TGF-β1 in BMDMs, but they were blocked by TG2 knockdown in vitro. The previous studies have reported that M2 macrophages can produce TGF-β1,[49] which contradicted our conclusions. This finding may be related to the various subtypes of M2 macrophages. M2 macrophages comprise four subtypes: M2a, M2b, M2c, and M2d. M2a secretes IL-10 and TGF-β; M2b secretes anti- and pro-inflammatory factors IL-10, IL-6, and IL-1β, and tumor necrosis factor-α; M2c secretes TGF-β and IL-10; and M2d secretes IL-4, IL-10, and IL-13.[50,51] Different subtypes can secrete various cytokines and chemokines and perform a variety of biological functions.[50] This outcome may explain the inhibition of TGF-β1/Smad3 signaling and the enhanced polarization of M2 macrophages in our study; similar results have also been reported in a previous study.[52] The effect of TG2 on M2 macrophage subtypes in MI must be further explored.
SUMMARY
The results reveal that TG2 inhibition ameliorated cardiac fibrosis post-MI. Its potential mechanism may induce M2 macrophage polarization and inactivate the TGF-β1/Smad3 pathway, which supports that TG2 may be a potential target in the therapy of MI.
AVAILABILITY OF DATA AND MATERIALS
The data of this study are available from the corresponding author on reasonable request.
ABBREVIATIONS
TG2: Transglutaminase 2
MI: Myocardial infarction
TGF-β: Transforming growth factor β
TGF-βR: TGF-β receptor
IL-10: Interleukin-10
VEGF: Vascular endothelial growth factor
Smad 2/3: Small mother against decapentaplegic 2/3
p-Smad3: phospho-Smad3
TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling
BMDMs: Bone marrow derived macrophages
LAD: Left anterior descending artery
LVEF: Left ventricular ejection fraction
LVFS: Left ventricular fraction shortening
LV vol: s: Left ventricular volume at end diastole
LVIDs: Left ventricular internal dimension in systole
KCl: Potassium chloride
BSA: Bovine serum albumin
FITC: Fluorescein isothiocyanate
DAPI: 4,6-diamino-2-phenyl indole
CD206: Macrophage mannose receptor
Arg-1: Arginase-1
F4/80: Epidermal growth factor-like module-containing
mucin-like hormone receptor-like 1
α-SMA: α-smooth muscle actin
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
AUTHOR CONTRIBUTIONS
QYC and AM: Designed the research; QYH and YRW: Performed the experiments; YRW and SJS: Analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by ethics committee of Huashan Hospital, Fudan University (No: 2020-JS-574), and all animal procedures complied with approved protocols. This study did not involve any human sample hence, consent to participate is not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This study is supported by the Clinical Research Project of Shanghai Municipal Health Commission (202140506).
References
- Ginkgolide A alleviates cardiac remodeling in mice with myocardial infarction via binding to matrix metalloproteinase-9 to attenuate inflammation. Eur J Pharmacol. 2022;923:174932.
- [CrossRef] [PubMed] [Google Scholar]
- Synthetic recovery of impulse propagation in myocardial infarction via silicon carbide semiconductive nanowires. Nat Commun. 2022;13:6.
- [CrossRef] [PubMed] [Google Scholar]
- Treatment of myocardial infarction with gene-modified mesenchymal stem cells in a small molecular hydrogel. Sci Rep. 2017;7:15826.
- [CrossRef] [PubMed] [Google Scholar]
- Rapid surface cooling by thermosuit system dramatically reduces scar size, prevents post-infarction adverse left ventricular remodeling, and improves cardiac function in rats. J Am Heart Assoc. 2015;4:e002265.
- [CrossRef] [PubMed] [Google Scholar]
- Myocardial fibrosis: Biomedical research from bench to bedside. Eur J Heart Fail. 2017;19:177-91.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac fibrosis: The fibroblast awakens. Circ Res. 2016;118:1021-40.
- [CrossRef] [PubMed] [Google Scholar]
- Global burden of cardiovascular diseases and risk factors, 1990-2019: Update from the GBD 2019 study. J Am Coll Cardiol. 2020;76:2982-3021.
- [CrossRef] [PubMed] [Google Scholar]
- Global cardiovascular diseases death rate prediction. Curr Probl Cardiol. 2023;48:101622.
- [CrossRef] [PubMed] [Google Scholar]
- The heart failure epidemic. Int J Environ Res Public Health. 2010;7:1807-30.
- [CrossRef] [PubMed] [Google Scholar]
- Transglutaminase 2 as a therapeutic target for neurological conditions. Expert Opin Ther Targets. 2021;25:721-31.
- [CrossRef] [PubMed] [Google Scholar]
- Role of transglutaminase 2 in cell death, survival, and fibrosis. Cells. 2021;10:1842.
- [CrossRef] [PubMed] [Google Scholar]
- TGM2 and implications for human disease: Role of alternative splicing. Front Biosci (Landmark Ed). 2013;18:504-19.
- [CrossRef] [PubMed] [Google Scholar]
- Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:699-707.
- [CrossRef] [PubMed] [Google Scholar]
- Imidazolium-based warheads strongly influence activity of water-soluble peptidic transglutaminase inhibitors. Eur J Med Chem. 2013;66:526-30.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis. 2018;9:613.
- [CrossRef] [PubMed] [Google Scholar]
- Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP synthesis. Cell Death Differ. 2006;13:1827-9.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammation as a treatment target after acute myocardial infarction. N Engl J Med. 2019;381:2562-3.
- [CrossRef] [PubMed] [Google Scholar]
- Efficacy of different drug-eluting stents and their influence on inflammation and prognosis in elderly patients with acute myocardial infarction. J Biol Regul Homeost Agents. 2021;35:1047-52.
- [Google Scholar]
- Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090.
- [CrossRef] [PubMed] [Google Scholar]
- Macrophage activation and polarization in post-infarction cardiac remodeling. J Biomed Sci. 2017;24:13.
- [CrossRef] [PubMed] [Google Scholar]
- New insights and novel therapeutic potentials for macrophages in myocardial infarction. Inflammation. 2021;44:1696-712.
- [CrossRef] [PubMed] [Google Scholar]
- CD16+ monocytes and skewed macrophage polarization toward M2 type hallmark heart transplant acute cellular rejection. Front Immunol. 2017;8:346.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic hepatitis C infection-induced liver fibrogenesis is associated with M2 macrophage activation. Sci Rep. 2016;6:39520.
- [CrossRef] [PubMed] [Google Scholar]
- Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13:3056-62.
- [CrossRef] [PubMed] [Google Scholar]
- Loss of Acta2 in cardiac fibroblasts does not prevent the myofibroblast differentiation or affect the cardiac repair after myocardial infarction. J Mol Cell Cardiol. 2022;171:117-32.
- [CrossRef] [PubMed] [Google Scholar]
- Sodium lactate accelerates M2 macrophage polarization and improves cardiac function after myocardial infarction in mice. Cardiovasc Ther. 2021;2021:5530541.
- [CrossRef] [PubMed] [Google Scholar]
- Caveolin-1 deletion exacerbates cardiac interstitial fibrosis by promoting M2 macrophage activation in mice after myocardial infarction. J Mol Cell Cardiol. 2014;76:84-93.
- [CrossRef] [PubMed] [Google Scholar]
- Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front Physiol. 2015;6:107.
- [CrossRef] [PubMed] [Google Scholar]
- Smad3 signatures in renal inflammation and fibrosis. Int J Biol Sci. 2022;18:2795-806.
- [CrossRef] [PubMed] [Google Scholar]
- M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol Int. 2017;41:960-8.
- [CrossRef] [PubMed] [Google Scholar]
- Myocyte enhancer factor 2A contributes to the TGF-β1-mediated cholangiocyte epithelial to mesenchymal transition and senescence in cholestatic liver fibrosis. Front Biosci (Landmark Ed). 2022;27:324.
- [CrossRef] [PubMed] [Google Scholar]
- Transglutaminases in wound healing and inflammation. Prog Exp Tumor Res. 2005;38:89-114.
- [CrossRef] [PubMed] [Google Scholar]
- Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids. 2004;26:387-404.
- [CrossRef] [PubMed] [Google Scholar]
- Matrix metalloproteinases and tissue inhibitor of matrix metalloproteinases expression by tubular epithelia and interstitial fibroblasts in the normal kidney and in fibrosis. Exp Nephrol. 1995;3:88-9.
- [Google Scholar]
- The role of transglutaminase in the rat subtotal nephrectomy model of renal fibrosis. J Clin Invest. 1997;99:2950-60.
- [CrossRef] [PubMed] [Google Scholar]
- Urinary matrix metalloproteinase activity in diabetic kidney disease: A potential marker of disease progression. Nephron Extra. 2012;2:219-32.
- [CrossRef] [PubMed] [Google Scholar]
- Matrix metalloproteinases: Their potential role in the pathogenesis of diabetic nephropathy. Endocrine. 2009;35:1-10.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac specific overexpression of transglutaminase II (G(h)) results in a unique hypertrophy phenotype independent of phospholipase C activation. J Biol Chem. 1999;274:21291-6.
- [CrossRef] [PubMed] [Google Scholar]
- Blocking TG2 attenuates bleomycin-induced pulmonary fibrosis in mice through inhibiting EMT. Respir Physiol Neurobiol. 2020;276:103402.
- [CrossRef] [PubMed] [Google Scholar]
- Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology. 2012;56:1117-28.
- [CrossRef] [PubMed] [Google Scholar]
- Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818-29.
- [CrossRef] [PubMed] [Google Scholar]
- Calhex231 ameliorates myocardial fibrosis post myocardial infarction in rats through the autophagy-NLRP3 inflammasome pathway in macrophages. J Cell Mol Med. 2020;24:13440-53.
- [CrossRef] [PubMed] [Google Scholar]
- The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549-74.
- [CrossRef] [PubMed] [Google Scholar]
- Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770-83.
- [CrossRef] [PubMed] [Google Scholar]
- Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47-64.
- [CrossRef] [PubMed] [Google Scholar]
- Transforming growth factor-β in tissue fibrosis. J Exp Med. 2020;217:e20190103.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64:526-35.
- [CrossRef] [PubMed] [Google Scholar]
- M2-like macrophage infiltration and transforming growth factor-β secretion during socket healing process in mice. Arch Oral Biol. 2021;123:105042.
- [CrossRef] [PubMed] [Google Scholar]
- Dissection of pro-tumoral macrophage subtypes and immunosuppressive cells participating in M2 polarization. Inflamm Res. 2024;73:1411-23.
- [CrossRef] [PubMed] [Google Scholar]
- Biochemical and molecular inducers and modulators of M2 macrophage polarization in clinical perspective. Int Immunopharmacol. 2023;122:110583.
- [CrossRef] [PubMed] [Google Scholar]
- PTEN inhibitor attenuates cardiac fibrosis by regulating the M2 macrophage phenotype via the PI3K/AKT/TGF-β/Smad 2/3 signaling pathway. Int J Cardiol. 2022;356:88-96.
- [CrossRef] [PubMed] [Google Scholar]




