


Translate this page into:
Analyzing histopathological aspects and cell populations in orbital inflammatory involvement in systemic diseases: A case series from the Rheumatologist’s perspective


*Corresponding author: Marco Zeppieri, Department of Ophthalmology, University Hospital of Udine, Udi ne, Italy. markzeppieri@hotmail.com
-
Received: ,
Accepted: ,
How to cite this article: Foti R, Foti R, Zeppieri M, Gagliano C. Analyzing histopathological aspects and cell populations in orbital inflammatory involvement in systemic diseases: A case series from the Rheumatologist’s perspective. CytoJournal. 2024;21:23. doi: 10.25259/Cytojournal_21_2024
Abstract
Orbital inflammatory disease (OID) comprises approximately 6% of orbital conditions, affecting individuals across all ages. The range of the primary orbital inflammation’s differential diagnosis is extensive, encompassing autoimmune disorders such as thyroid diseases, vasculitis, sarcoidosis, connective tissue diseases, immunoglobulin G4-related disease (IgG4-RD), and giant cell myositis, whereas secondary causes span from infections to drug-induced causes. Analyzing histopathological aspects and cell populations could enhance our comprehension of the etiology of orbital inflammatory involvement in systemic diseases such as IgG4-RD. We present a series of four patients from our Rheumatology clinic, each with distinct systemic diseases, illustrating diverse manifestations of OID. This series was conducted to facilitate discussions and diagnoses of challenging cases of OID in a rheumatologic setting. The difficulty in the differential diagnosis arises from the extensive range of structures involved, resulting in a significant variation of clinical manifestations. Furthermore, the lack of definitive diagnostic laboratory tests and, often, histological findings add to the complexity. OID poses diagnostic challenges with variable clinical manifestations and overlapping imaging findings. As a diagnosis of exclusion, a comprehensive evaluation is crucial, often necessitating an orbital biopsy for confirmation. Collaborative efforts among specialists are essential for managing these intricate cases.
Keywords
Orbit
Orbital inflammation
Eye
Autoimmune disease
Vasculitis

INTRODUCTION
Orbital inflammatory disease (OID) constitutes 6% of orbital diseases, presenting with diverse findings based on inflammation, elevated orbital pressure, and compression effects. Common signs include conjunctival injection, chemosis, eyelid swelling, proptosis, diplopia, pain with eye movement, ophthalmoplegia, and impaired vision.[1] Primary orbital inflammation’s differential diagnosis spans idiopathic inflammatory disease, autoimmune or autoimmune-like disorders (thyroid diseases, vasculitis, sarcoidosis, connective tissue diseases, immunoglobulin G4-related disease (IgG4-RD), and giant cell myositis). Secondary orbital inflammation stems from systemic or local inflammatory conditions, and associated conditions (infections, neoplasms, congenital malformation, trauma, Erdheim-Chester, and histiocytosis X). This series reports four cases with diverse systemic diseases and OID manifestations [Table 1]. OID often presents as a manifestation of systemic autoimmune or autoinflammatory conditions, and rheumatologists play a crucial role in diagnosing and managing these complex cases.
| Case | Age | Sex | Diagnosis | Histological findings | Treatment |
|---|---|---|---|---|---|
| Case 1 | 53 years old | Male | Idiopathic orbital inflammation | Periductal and perivascular lymphocytic infiltrates of CD3+LAT+ T cells, increased connective tissue, without a significant increase of IgG4+ plasma cells | Methotrexate (17.5 mg/week) + prednisone |
| Case 2 | 48 years old | Male | Sjögren’s syndrome associated high-grade non-Hodgkin peripheral lymphoma | Biopsy of neck soft tissues in February 2019 revealed nodular interstitial, perivascular, perineural infiltrates of lymphoid (B cells, mostly T cells), histiocytic, plasmacellular elements; high number of plasma cells (k>l) and activated histiocytic cells (CD68+ CD163+). (biopsy of neck soft tissue) | R-CHOP |
| Case 3 | 42 years old | Female | Granulomatosis with polyangiitis | Inflammatory necrotic tissue with granulomatosis and autoreactive IgG4+ B cells | Cyclophosphamide therapy + low-dose prednisone Rituximab |
| Case 4 | 26 years old | Female | Idiopathic orbital inflammation | Degenerative processes, fibrotic involution, massive T cell infiltrate. |
Azathioprine 50 mg/die + prednisone |
R-CHOP: Rituximab - Cyclophosphamide, doxorubicin, vincristine and prednisolone
OID is not isolated but is often associated with systemic diseases, including autoimmune disorders such as thyroid diseases, vasculitis, sarcoidosis, connective tissue diseases, IgG4-RDs, and giant cell myositis. Rheumatologists specialize in the diagnosis and management of systemic autoimmune and inflammatory conditions. This case series showcases how OID can be a clinical manifestation of these underlying systemic diseases. Rheumatologists should be trained to deal with the complexities of diagnosing autoimmune and inflammatory conditions, and OID poses unique diagnostic challenges due to its variable clinical manifestations and overlapping imaging findings.
The histopathological analysis is a crucial aspect in rheumatology, to enhance the understanding of OID etiology. Rheumatologists often rely on histopathological findings to confirm diagnoses and tailor treatment plans. Rheumatologists play a key role in interpreting histopathological results and integrating them into the broader context of systemic autoimmune diseases. Treatment includes the use of immunosuppressants and steroids. The involvement of rheumatologists in OID cases highlights the importance of immunomodulation in the management of these conditions, as the immune system plays a central role in both rheumatologic disorders and OID. Furthermore, interdisciplinary care is essential to address the complexities of OID in the context of systemic diseases.
CASE SERIES
Case 1
A 53-year-old male farmer, presenting in April 2017 with left orbital swelling and occasional diplopia, underwent eye sonography, revealing extrinsic muscle thickening with edema, excluding diplopia. Orbital magnetic resonance imaging (MRI) in September 2017 showed severe thickening of the superior right rectus muscle with marked enhancement after gadolinium. Routine tests were normal except for slightly elevated IgG4 (1.80 g/L) and slightly increased chitotriosidase values of 44.57 nmol/mL (normal value <25). All other routine laboratory tests were normal including markers of inflammation, angiotensin-converting enzyme (ACE), Bence–Jones, thyroid functionality, and autoimmunity. Occasionally, hypereosinophilia was reported. Oral prednisone (max 50 mg/day) was prescribed in September 2017, with azathioprine added in June 2018 due to low tolerability. A December 2018 flare led to an orbital MRI in January 2019, showing optic nerve dislocation and muscle swelling [Figure 1]. Positron emission tomography (PET) imaging revealed no abnormalities. A March 2019 orbital tissue biopsy revealed two periductal and perivascular lymphocytic infiltrates of CD3+ LAT+ T cells, increased connective tissue, without a significant increase of IgG4 positive plasma cells [Figure 2], leading to an idiopathic orbital inflammation (IOI) diagnosis. Methotrexate (17.5 mg/week) initiated in March 2019 resulted in clinical improvement, and prednisone was tapered to 25 mg/day. Follow-up examinations showed no pain, no edema, bilateral hypertrophic lacrimal glands, normal extrinsic ocular motricity, and no diplopia. Orbital MRI demonstrated reduced inflammatory tissue, regular optical nerve course, and normal orbital adipose tissue [Figure 3]. As of the last follow-up in November 2021, the patient remains in remission, continuously taking methotrexate at 10 mg/week.

- Case 1 magnetic resonance imaging with contrast scan in December 2018. (a) A red arrow is pointing to tissue (isointense to muscles in T1, hyperintense in T2-short-tau inversion recovery (STIR), and with post-contrast enhancement) molds and does not appear dissociable from the upper complex, obliterating both extraconal and intraconal fat. (b) A red arrow is pointing to imprint of the proximal portion of the optic nerves. (c) A red arrow is showing that all extraocular eye muscles appear swollen and hyperintense in STIR, indicative of edema.
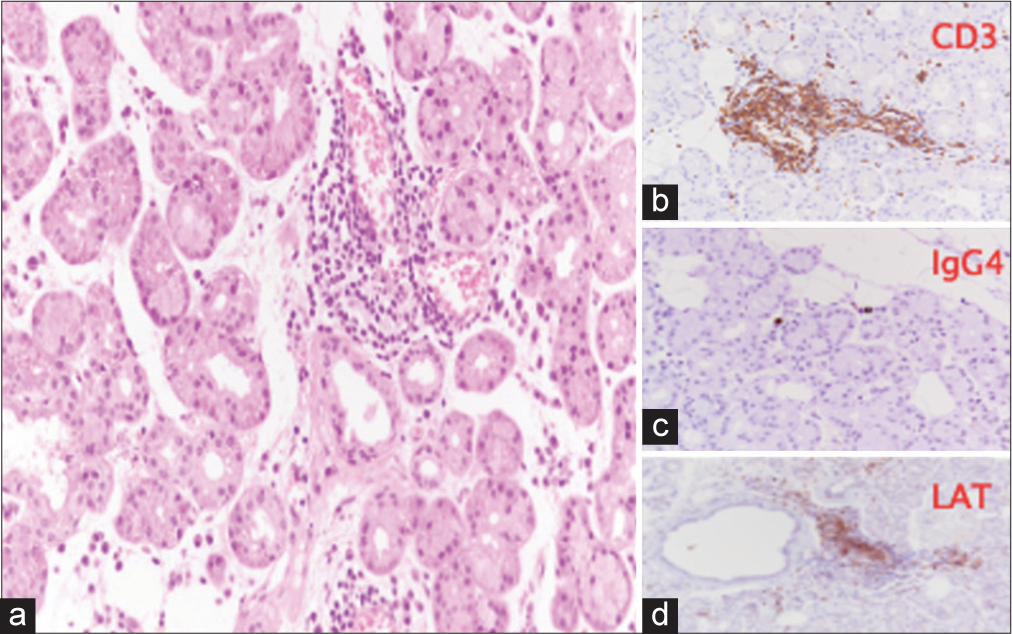
- Case 1 biopsy of the right orbital tissues, performed in March 2019. (a) Lobules of lacrimal gland with >two foci of periductal and perivascular lymphocytic infiltration and minor interstitial infiltrate, consisting of CD3+ LAT+ T lymphocytes (b & d), without a significant increase in immunoglobulin G4-positive plasma cells (c). (Hematoxylin and Eosin (H&E) stain, ×20 magnification). (CD3: Cluster of Differentiation 3, IGg4: Immunoglobulin G4, LAT: Linker for activation of T cells.)
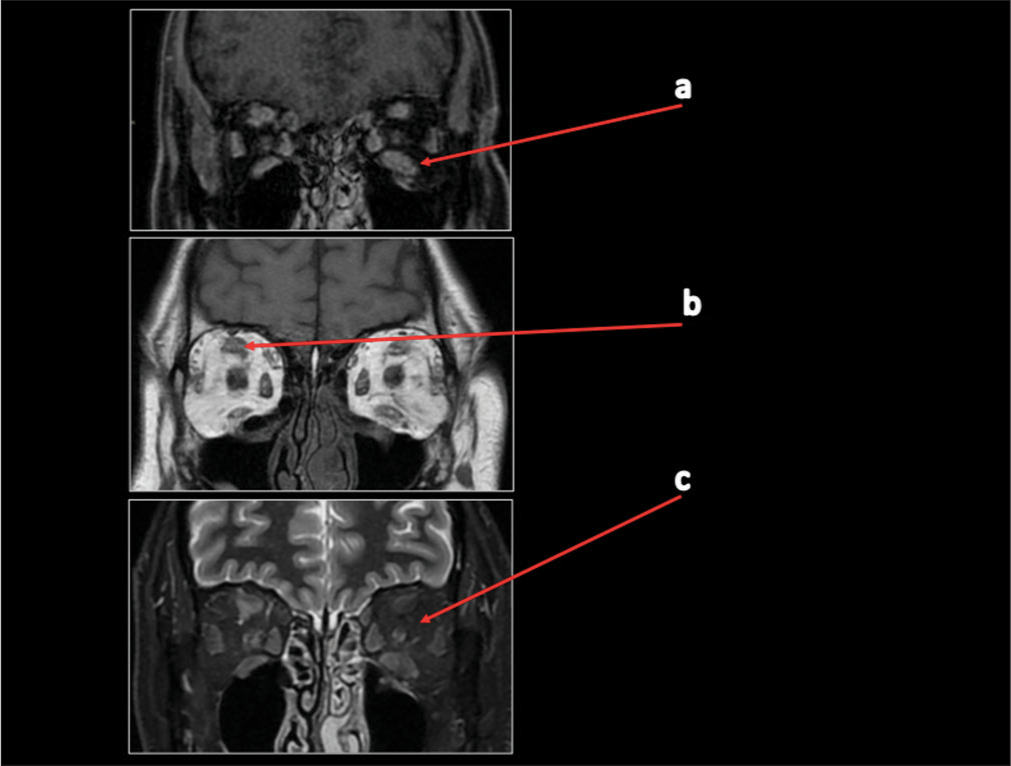
- Case 1 magnetic resonance imaging with contrast scan in November 2021. (a) A red arrow is pointing to volumetric reduction of tumefactive alterations in the extrinsic muscles with modest contrast enhancement of the examined muscle structures. (b) A red arrow is pointing to a slight decrease in the size of the solid tissue that was previously inseparable from the complex of the upper right rectus. (c) A red arrow is showing that both optic nerves and intra-orbital adipose tissue exhibit normal signal, morphology, and course.
Case 2
In July 2018, a 48-year-old male volcanological guide presented prolonged fever, painful salivary gland swelling, and diffuse arthralgias. August 2018 abdominal sonography confirmed splenomegaly; computed tomography (CT) scans revealed multiple lymphadenopathy sites. Laboratory results indicated elevated C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). PET scan showed increased metabolic activity in lymph nodes. Salivary gland histopathology was consistent with Sjögren’s Syndrome. Nonsteroidal anti-inflammatory drugs (NSAIDs) and paracetamol reduced salivary gland swelling, occasional fever, and no joint involvement by November 2018. In December 2018, erythematous cutaneous nodules appeared with daily fever, arthralgia, increased salivary gland swelling, right neck lymphadenopathy, right face hypoesthesia, and xerophthalmia. January 2019 worsened symptoms, including right orbit pain, ocular elevation deficit, diplopia, cough, mild dyspnea, and night sweats. February 2019 cranial CT revealed lymphadenopathies, parotid swelling, and right inferior rectus muscle enlargement [Figure 4]. Blood tests indicated leukocytosis and elevated inflammation indices. A biopsy of neck soft tissues in February 2019 revealed nodular interstitial, perivascular, and perineural infiltrates of lymphoid (B cells, mostly T cells), histiocytic, and plasma cellular elements. B cells (CD20+ bcl2+/− bcl6−/+ CD10−/+ CD5− CD43−) and T cells (CD3+ CD43+ bcl2+/− CD5+ CD4>CD8) did not show a clear neoplastic pattern; high number of plasma cells (k>L) and activated histiocytic cells (CD68+ CD163+). Medium fraction of growth (Ki-67/Mib-1: 20–25%). Deoxyribonucleic acid (DNA) analysis for immunoglobulin (Ig) H gene rearrangements showed Igs heavy chains FR2-JH consistent with the presence of a clonal/oligoclonal population. March 2019 orbital MRI revealed significant swelling of the right inferior rectus muscle with unilateral exophthalmos. Thyroid function was normal. April 2019 lymph node biopsy diagnosed a high-grade non-Hodgkin peripheral lymphoma, leading to referral to oncohematological care and R-CHOP treatment.
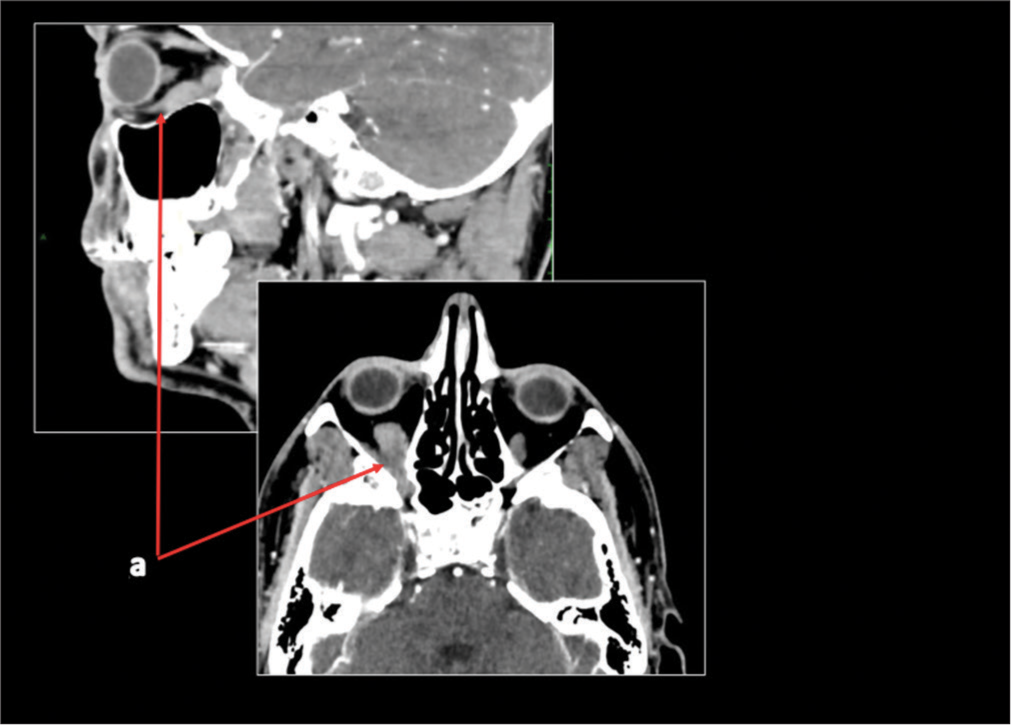
- Case 2 computed tomography scan in February 2019. A red arrow is pointing to a volumetric increase of the inferior rectus muscle of the right orbit (measuring 12 mm) compared to the left side (9 mm).
Case 3
In September 2017, a 42-year-old female developed bilateral mucopurulent rhinosinusitis and sub-acute mucopurulent otitis media. Laboratory tests revealed cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA) positivity (1:40, antiPR3 29 UI/L) and elevated inflammation indices. Suspecting granulomatosis with polyangiitis, she received prednisone 25 mg with tapering and azathioprine 50 mg twice daily, leading to symptom improvement and subsequent therapy cessation due to complete remission.
In March 2018, she experienced left ocular pain, diplopia, proptosis, left strabismus, facial pain, bilateral maxillary heaviness, and nasal discharge. Visual impairment and left eye abduction deficit developed. Bilateral functional endoscopic sinus surgery (FESS) in April 2018 included left optic nerve decompression, with inconclusive biopsy. High-dose intravenous steroids resulted in ophthalmoplegia and fixed mydriasis in the left eye.
April 2018, orbital computed tomography (CT) and magnetic resonance imaging (MRI) revealed thickening and edema of rectus muscles (inferior and medial) and left ocular nerve compression. May 2018 cerebral MRI showed worsening periorbital edema and conjunctival chemosis, with enhancement in the middle cranial fossa. A June 2018 rheumatology admission confirmed previous findings, indeed the patient exhibited pronounced left exophthalmos accompanied by thickening of the optic nerve, retrobulbar fat tissue hyperintensity, and evidence of edema and inflammation in the left ethmoidal cells, sphenoid sinus, and frontal sinus. These observations were further accentuated following gadolinium administration and were consistent with the ocular manifestation of granulomatosis with polyangiitis.
Revision of a previous biopsy indicated inflammatory necrotic tissue with granulomatosis and autoreactive IgG4-positive B cells. Marked left exophthalmos persisted [Figure 5], with orbital magnetic resonance (MR) showing optical nerve thickening, retrobulbar fat tissue hyperintensity, and left ethmoidal, sphenoid, and frontal sinus inflammation, compatible with granulomatosis with polyangiitis [Figure 6].
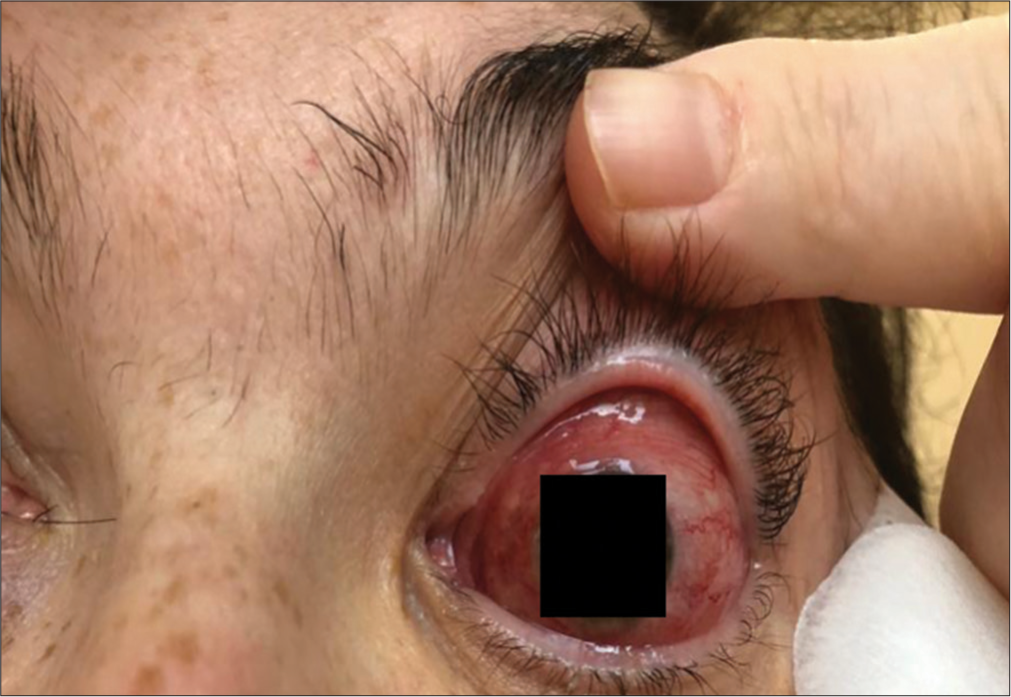
- Clinical presentation of Case 3 in June 2018.
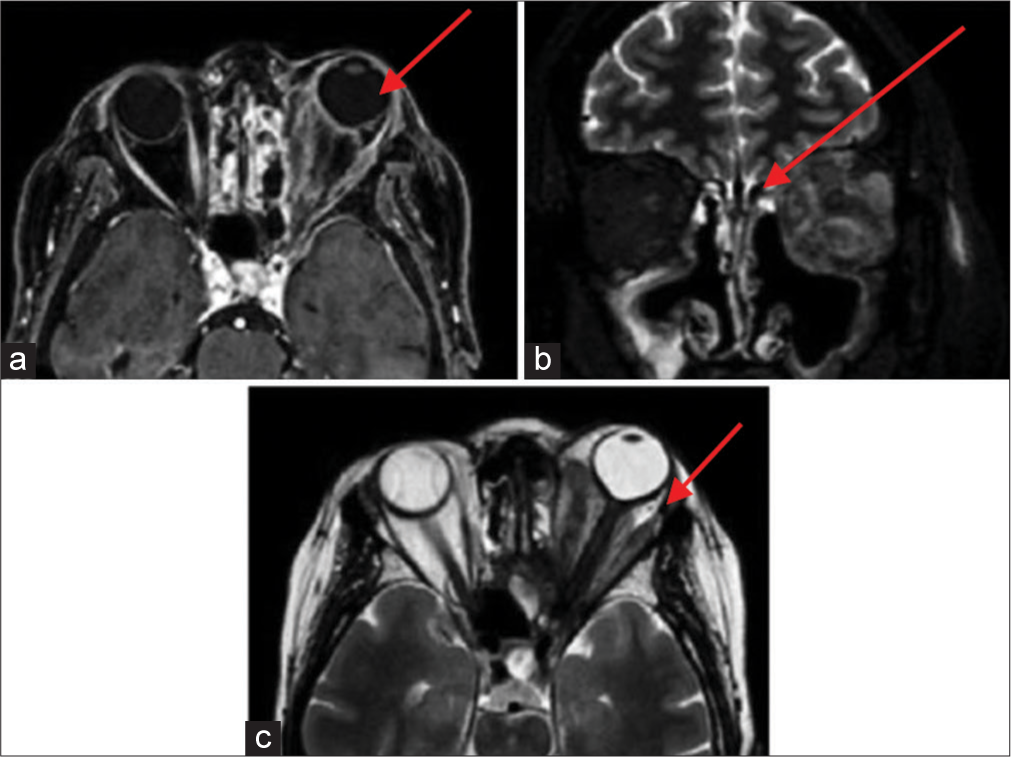
- Case 3 magnetic resonance imaging with contrast scan in June 2018. (a) A red arrow is pointing to a marked condition of left exophthalmos, accompanied by a significant volumetric increase in the extrinsic ocular musculature, the optic nerve in all assessable segments with liquid distension of the peri-optic sheath. There is pronounced hyperintensity in signal within the retrobulbar adipose tissue, along with soft-tissue edema. (b) A red arrow is pointing to evidence of inflammatory tissue involving the ethmoidal cells on the left side, the sphenoidal sinus, and the frontal sinuses, particularly on the left. (c) A red arrow is pointing to a notable volumetric increase visible following intravenous gadolinium administration in all previously mentioned structures consistent with the clinical inquiry, suggesting an ocular manifestation of Wegener’s granulomatosis.
Granulomatosis with polyangiitis diagnosis led to pulse cyclophosphamide therapy with low-dose prednisone. Subsequent rituximab therapy resulted in progressive improvement. November 2021 orbital MR demonstrated decreased inflammatory tissue and orbital fat tissue.
Case 4
A 26-year-old female student, in 2011, presented with sudden burning pain and conjunctival hyperemia in her right eye. A biopsy confirmed idiopathic myositis, treated successfully with steroids. In 2016, a left eye flare occurred with negative antinuclear antibodies (ANA), thyrotropin receptor antibodies (TRABs), normal creatine kinase (CK), and inflammation markers. Oral steroids (max 25 mg/day) were initiated. Recurrent right orbital pain led to occasional steroid and Non-Steroidal Anti-Inflammatory Drugs (NSAID) use. In June 2020, a right eye flare showed, on ocular examination, mild enophthalmos in the right eye, alongside relative exophthalmos in the left eye, accompanied by pain on palpation of the superior orbital regions. MRI findings revealed severe thickening of the medial rectus muscle, along with mild thickening of the lateral rectus and superior oblique muscles of the eye [Figure 7]. Once again, oral steroid therapy proved effective in managing the condition. January 2021 brought a left eye flare, worsening diplopia, and medial rectus muscle involvement. Pulse steroids (500 mg intravenous for 4 days) were started. A biopsy of the left eye’s lateral rectus muscle showed degenerative processes, fibrotic involution, and massive T cell infiltrate [Figure 8]. A diagnosis of IOI was made. In May 2021, azathioprine 50 mg with prednisone 10 mg/day was initiated resulted in rapid improvement with no flare-ups up to the November 2021 follow-up.
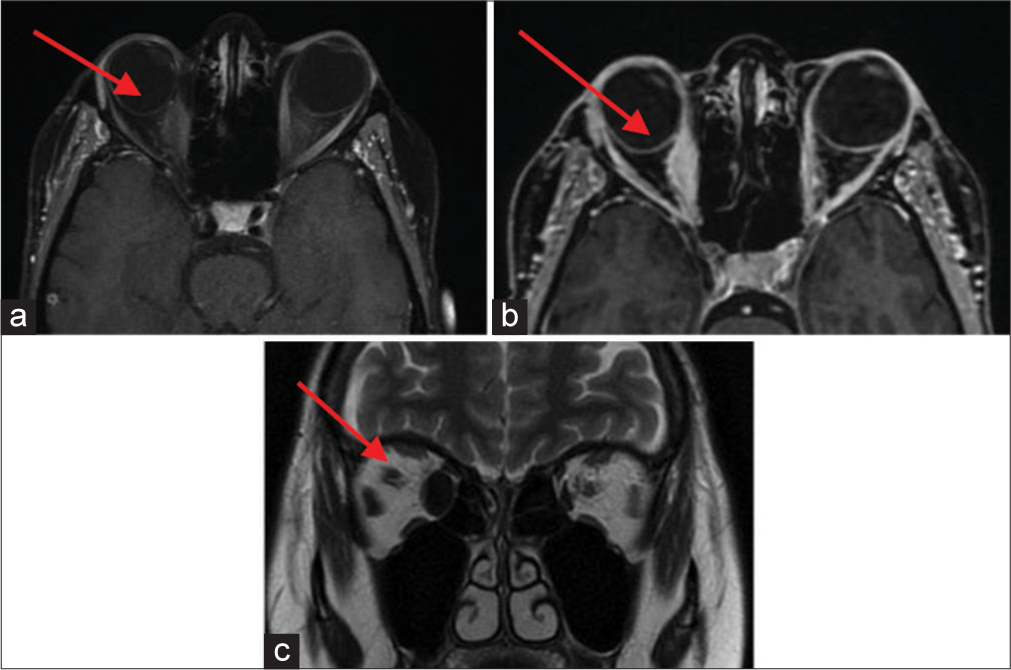
- Case 4 magnetic resonance imaging with contrast scan in August 2020. (a) A red arrow is pointing to a fusiform thickening of the right medial rectus muscle (10 mm). (b) A red arrow is pointing to a slight thickening in the right lateral rectus muscle (4 mm compared to 2.3 mm at the left side) and the right superior oblique muscle (4.2 mm on the right compared to 2.3 mm at the left side). (c) A red arrow is pointing to evidence of a slightly sinuous course in the intraorbital segment of the optic nerve at the left side. Both optic nerves exhibit a regular signal.
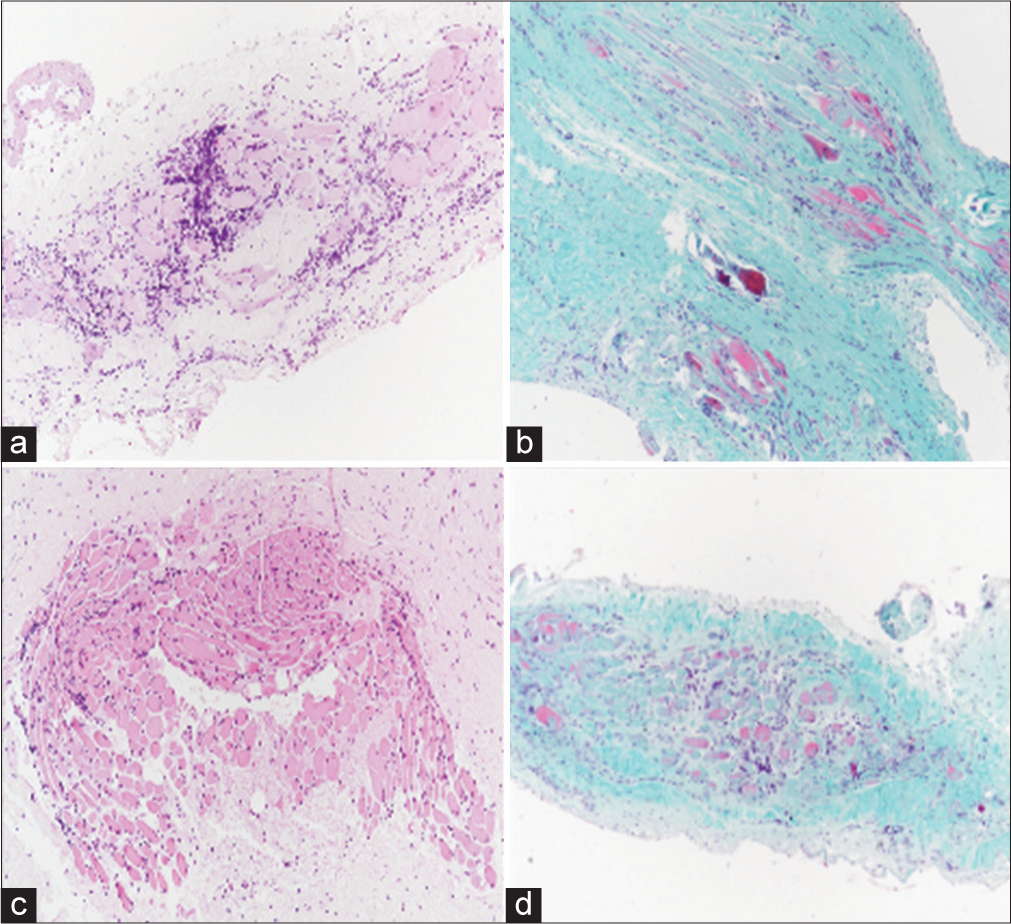
- Case 4 biopsy of the lateral rectus muscle of the left eye, performed in May 2021. (a-d) are illustrating different parts of soft tissues and bundles of striated muscle that show degenerative-regressive phenomena of muscle fibrocells, collagen fibrotic dissociation, and significant T-cell lymphocytic infiltration. (Hematoxylin and eosin (H/E) stained, ×20 magnification).
DISCUSSION
This series delves into complex OID cases in rheumatology. Clinical manifestations and inconclusive findings pose a challenging differential diagnosis. In a retrospective case series of 24 patients with biopsy-proven non-specific OID, involvement varied: Lacrimal gland (54.2%), extraocular muscles (50%), orbital fat (75%), sclera (4.2%), optic nerve (20.8%), and other structures (8.3%).[2] The histopathologic spectrum is often non-diagnostic, reflecting diverse presentations such as diffuse polymorphous infiltrate, lymphoid, granulomatous, sclerosing, eosinophilic, or vasculitic inflammation.[3] Biopsy is crucial for resolving those doubtful cases where diagnostic certainty is needed to promptly initiate therapy. Therefore, it is suggested to proceed with a biopsy of the area as soon as possible, even before the initiation of therapy, if possible. However, a delay in performing the biopsy should not delay the initiation of therapy, especially in the presence of strong clinical suspicion of a systemic and progressive disease. The biopsy should be performed at a specialized center, with a well-trained team for execution. It would be preferable for the team to also include a plastic surgeon, whose expertise could ensure the use of the least invasive and most skin-preserving technique possible. Rheumatologists, ophthalmologists, radiologists, and surgeons should hold pre-operative discussion meetings to pinpoint the area of greatest interest for biopsy based on imaging evidence, and efforts should be made to employ minimally invasive techniques. Indeed, the orbital area is a very delicate zone that can be adversely affected by poor scarring if not treated carefully. Typically, trajectory-guided procedures are the most commonly utilized and can be pre-planned for individual cases. Minimally invasive procedures are usually suitable for obtaining biopsies of the orbit in the majority of cases. However, for certain patients, a conventional, non-minimally invasive lateral orbitotomy may be necessary.
Case 1
In an initial IgG4 disease diagnosis seemed likely based on clinical presentation, MRI, and slightly elevated IgG4. However, the biopsy excluded IgG4-positive cells, leading to an IOI diagnosis. IOI is characterized by histological findings such as lymphocytic, granulocytic, and sometimes histiocytic infiltrates, organized into lymph follicles with germinal centers, increased connective tissue with edema and fibrosis, and early destruction of lacrimal gland acini or muscle fibers. Typical symptoms include painful pseudoptosis, chemosis, exophthalmos, and impaired ocular motility. Comprehensive laboratory exams and serological diagnostics, including kidney and thyroid values, Anti-neutrophil cytoplasm antibodies (ANCA), Angiotensin converti enzyme (ACE), rheumatoid factors, and IgG4 serum level, are useful. The therapeutic approach includes NSAIDs, steroids, and immunosuppressants.[4]
Case 2, posed a diagnostic challenge with transient salivary gland manifestations. Histology supported Sjögren’s Syndrome. Orbital involvement, lacking specific features, is established in Sjögren’s syndrome,[5] with lymphoma as a severe complication.[6] In our case, essential histology confirmed a not otherwise specified lymphoma, verified by DNA analysis.
Case 3, suggestive of granulomatosis with polyangiitis, initially indistinguishable from IgG4 disease, underwent histology and revealed characteristic features. IgG4-RD is systemic, impacting various organs (pancreas, bile ducts, kidneys, lacrimal and salivary glands, and lymph nodes). Orbital manifestations involve painless lacrimal gland swelling, nerve-induced diplopia without pain, and enlarged lacrimal glands with orbital fatty tissue. Imaging depicts diffuse lacrimal gland enlargement, lateral muscle involvement, infraorbital nerve swelling, and often ipsilateral paranasal sinus involvement.[7] Histology showcases lymphoplasmacellular infiltration with obliterating phlebitis and eosinophilic inflammation. Criteria encompass >40% IgG4+ plasma cells, IgG4/IgG ratio >40%, or >50 IgG4+ cells/high-power field (×400). Laboratory findings show increased IgG4 serum levels but can be non-specific, especially in limited orbital involvement.[8] However, our patient did not meet IgG4 disease criteria due to normal serum IgG4 levels. The biopsy revealed granulomatosis with polyangiitis features, including vasculitis and inflamed necrotic tissue. In granulomatosis with polyangiitis, orbital involvement occurs in 15–20% of cases, either de novo or secondary to sinus disease, causing symptoms such as pain, diplopia, and decreased vision, with signs such as conjunctival injection, proptosis, and ophthalmoplegia. CT with contrast shows hyperintense lesions, obliteration of tissue planes, and bony erosion. On MRI, lesions are hypointense to orbital fat in T1 and T2, enhancing with gadolinium.[9]
Case 4 had a diagnosis of idiopathic orbital myositis from the first biopsy with a peculiar rate of remission and flare periods. Initial response to steroids is usual and has been already discussed in the first case as well as the need to use immunosuppressant agents for frequent or more severe flares.[10]
This short case series can be of potential clinical use and provide significance and contribution in a routine clinical setting. It offers an understanding of the diagnostic, histopathological, and therapeutic aspects of OID in the context of systemic diseases, with real-world cases providing valuable insights for both clinicians and researchers. The article highlights the diagnostic challenges associated with OID, emphasizing the complexity of differential diagnosis due to the varied clinical manifestations and overlapping imaging findings. This information is crucial for clinicians in rheumatology, providing insights into the intricacies of identifying OID in the context of systemic diseases. The focus on analyzing histopathological aspects and cell populations in OID contributes to a deeper understanding of the disease’s etiology, especially in the context of systemic diseases such as IgG4-RDs. It is important to stress the value of comprehensive evaluation, including orbital biopsy, to confirm diagnoses and address the lack of definitive diagnostic laboratory tests.
Case series diversity is also an important issue. The inclusion of a series of four diverse cases, each with distinct systemic diseases and OID manifestations, adds significant value. This diversity provides a comprehensive view of how OID can manifest in different systemic conditions, ranging from IOI to lymphoma, granulomatosis with polyangiitis, and idiopathic orbital myositis.
The article discusses therapeutic approaches for each case, detailing the treatments administered and their outcomes. The long-term follow-up information, such as remission, improvement, or flare-ups, contributes valuable insights into the effectiveness of different therapeutic strategies in managing OID associated with systemic diseases. Emphasizing the need for interdisciplinary collaboration, the article underscores the importance of collaborative efforts among specialists, particularly in rheumatology, to manage complex OID cases effectively. This highlights the necessity of a holistic approach involving various medical disciplines.
The article serves an educational purpose by providing a comprehensive review of the histopathological spectrum of OID, presenting it in a clinical context. It contributes to the existing literature by delving into the complexities of OID and its manifestations, offering a resource for researchers, clinicians, and educators. The detailed descriptions of cases and their outcomes have potential implications for both clinical practice and further research. Clinicians may find the presented cases relevant for improving their diagnostic and therapeutic approaches, whereas researchers may identify gaps in knowledge that warrant further investigation.
SUMMARY
Idiopathic orbital disease (IOD) presents both diagnostic and therapeutic challenges, involving a variety of orbit structures with differing manifestations. This diversity offers a comprehensive insight into the various ways IOD can manifest in different systemic conditions, ranging from idiopathic orbital inflammation (IOI) to lymphoma, granulomatosis with polyangiitis, and idiopathic orbital myositis.
Therapeutic approaches may vary for each case, and it is crucial to gather diverse perspectives on the effectiveness of different therapeutic strategies in managing IOD associated with systemic diseases. As a diagnosis of exclusion, a biopsy to rule out other orbital conditions is essential. Collaboration among multiple specialists is key to better understanding these diseases, tailoring therapy to individual patients, taking into account comorbidities and circumstances, and ultimately aiding the patient in their long-term healing process and follow-up care.
Continued study, research, and discussion are necessary for further insights and publications on these complex cases.
AVAILABILITY OF DATA AND MATERIALS
The authors confirm that the data supporting the findings of this study are available within the article.
ABBREVIATIONS
ANCA: Anti-neutrophil cytoplasm antibodies
ACE: Angiotensin converting enzyme
IOI: Idiopathic orbital inflammation
IgG4-RD: Immunoglobulin G4-related disease
OID: Orbital inflammation disease
AUTHOR CONTRIBUTIONS
RF and CG: Concept and writing; RF: Data collection, analysis and writing draft and editing the draft; MZ: Data interpretation, supervision, review and editing of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was granted exemption by the Ethics Committee as this is a case series made retrospectively and without identifiers. The authors certify that they have obtained all appropriate patient consent.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
SUPPLEMENTARY MATERIAL
Supplementary material associated with this article can be found doi: 10.25259/Cytojournal_21_2024
FUNDING
Not applicable.
References
- Orbital inflammatory disease: A diagnostic and therapeutic challenge. Eye (Lond). 2006;20:1196-206.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic orbital inflammatory syndrome: Clinical features and treatment outcomes. Br J Ophthalmol. 2007;91:1667-70.
- [CrossRef] [PubMed] [Google Scholar]
- Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery lecture, part 1. Ophthalmology. 2004;111:997-1008.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic orbital inflammation: Review of literature and new advances. Middle East Afr J Ophthalmol. 2018;25:71-80.
- [CrossRef] [PubMed] [Google Scholar]
- Orbital myositis and primary Sjögren syndrome. J Rheumatol. 2015;42:1536-7.
- [CrossRef] [PubMed] [Google Scholar]
- Primary Sjögren's syndrome is associated with increased risk of malignancies besides lymphoma: A systematic review and meta-analysis. Autoimmun Rev. 2022;21:103084.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical perspectives on IgG4-related disease and its classification. Annu Rev Med. 2022;73:545-62.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic criteria for IgG4-related ophthalmic disease. Jpn J Ophthalmol. 2015;59:1-7.
- [CrossRef] [Google Scholar]
- Updates of ocular involvement in granulomatosis with polyangiitis. Graefes Arch Clin Exp Ophthalmol. 2023;261:1515-23.
- [CrossRef] [PubMed] [Google Scholar]
- Orbital tumors and inflammatory disorders: Diagnosis and management. Int Ophthalmol Clin. 2018;58:181-95.
- [CrossRef] [PubMed] [Google Scholar]




