


Translate this page into:
Embryonic lethal abnormal vision like 1-stabilized histone deacetylase 6 promotes hepatic stellate cell activation to accelerate liver fibrosis progression through ribosomal protein S5 downregulation

*Corresponding author: Qin Wang, Department of Clinical Medicine, Gansu Second People’s Hospital Northwest University Affiliated Hospital for Nationalities, Gansu, China. Department of School of Medicine; Hainan University of Science and Technology, Hainan, China wodongchongxiacao@163.com
-
Received: ,
Accepted: ,
How to cite this article: Wang Q, Zhang W, Wang J, Zhang L, Qiu Y, Cheng Y. Embryonic lethal abnormal vision like 1-stabilized histone deacetylase 6 promotes hepatic stellate cell activation to accelerate liver fibrosis progression through ribosomal protein S5 downregulation. CytoJournal. 2025;22:30. doi: 10.25259/Cytojournal_221_2024
Abstract
Objective
Histone deacetylase 6 (HDAC6) has been confirmed to participate in the regulation of liver fibrosis (LF) progression. This study aims to explore the role and mechanism of HDAC6 in the LF process.
Material and Methods
Serum samples were collected from liver cirrhosis (LC) patients and normal healthy individuals. Human hepatic stellate cells (HSC; LX-2) were stimulated with transforming growth factor β1 (TGF-β1) to mimic LF cell models. The levels of HDAC6, ribosomal protein S5 (RPS5), embryonic lethal abnormal vision like 1 (ELAVL1), and fibrosis-related markers were determined by quantitative real-time polymerase chain reaction or western blot. Cell proliferation and invasion were detected using cell counting kit 8 assay, 5-ethynyl-2’-deoxyuridine assay, and Transwell assay. The contents of inflammatory factors were examined using enzyme-linked immunosorbent assay. Furthermore, co-immunoprecipitation and RNA immunoprecipitation assays were performed to assess the interaction between HDAC6 and RPS5 or ELAVL1. The effect of ELAVL1 knockdown on HDAC6 mRNA stability was evaluated using Actinomycin D treatment assay.
Results
HDAC6 showed increased expression in LC patients. The knockdown of HDAC6 reduced TGF-β1-induced LX-2 cell proliferation, invasion, fibrosis, and inflammation. Moreover, HDAC6 reduced the acetylation of RPS5, and RPS5 knockdown reversed the inhibition effect of si-HDAC6 on TGF-β1-induced LX-2 cell proliferation, invasion, fibrosis, and inflammation. Meanwhile, ELAVL1 interacted with HDAC6 to stabilize its mRNA, thus inhibiting RPS5 expression.
Conclusion
Our data revealed that ELAVL1-stabilized HDAC6 promoted TGF-β1-induced HSC activation by repressing RPS5 acetylation, thus providing a novel target for alleviating LF progression.
Keywords
Embryonic lethal abnormal vision like 1
Fibrosis
Histone deacetylase 6
Liver cirrhosis
Ribosomal protein S5

INTRODUCTION
Liver disease is a major global public health problem, and according to statistics, about 2 million people worldwide die from liver diseases each year, of which liver cirrhosis (LC) accounts for about half of the deaths.[1] Based on the principles of hepatocrinology, liver diseases have a complex bidirectional relationship with endocrine dysfunction, and LC can cause endocrine disruption due to toxin accumulation and protein synthesis disruption.[2,3] LC is a common chronic liver disease with the main pathological process of liver fibrosis (LF).[4] The progression of LF can aggravate the damage to liver structure and function, leading to LC and organ failure.[5,6] It has been reported that the main source of extracellular matrix is the activated hepatic stellate cell (HSC) and that the inhibition of HSC activation is an available strategy for LF therapy.[7] Therefore, exploring the molecular mechanisms affecting HSC activation may provide new ideas for alleviating the progression of LF.
Histone acetylation is an epigenetic modification involved in various biological processes and is performed by histone acetyltransferases and histone deacetylases (HDACs).[8] Histone deacetylase 6 (HDAC6), a class IIB deacetylase among HDACs with zinc-dependent isoforms, is a key regulator of human diseases.[9] HDAC6 is also involved in physiological and pathological processes through the deacetylation of non-histone proteins.[9] A study reported elevated HDAC6 expression in acetaminophen-induced liver injury cell models and the potential amelioration of liver injury by HDAC6 inhibitors through the suppression of cell oxidative stress.[10] Wang et al. found elevated HDAC6 protein levels in the serum of LF rats, concluding that the HDAC inhibitor suberoylanilide hydroxamic acid alleviated the LF process by reducing HDAC6 expression.[11] However, whether HDAC6 mediates LF progression by regulating the activation of HSC requires further investigation.
Ribosomal protein S5 (RPS5) is one of the important members of the RP family and a main component of the eukaryotic ribosome.[12] RPS5 has been confirmed to be related to the development of many human diseases, such as pulmonary arterial hypertension and hepatocellular carcinoma (HCC).[13,14] A previous study suggested that RPS5 upregulation could relieve LF progression by preventing HSC activation,[15] thus confirming the negative role of RPS5 in LF. Embryonic lethal abnormal vision like 1 (ELAVL1, also known as HuR) is a typical RNA binding protein. Ge et al. found that ELAVL1 activated transforming growth factor β1 (TGF-β1)-induced HSC and accelerated the LF process by binding to sphk1 mRNA.[16] Through database prediction and experimental verification, we found that ELAVL1 interacted with HDAC6 to stabilize its expression and that HDAC6 might deacetylate RPS5. However, it remains unknown whether ELAVL1-mediated HDAC6 regulates HSC activation by mediating the acetylation of RPS5.
In the present study, we hypothesized that ELAVL1-stabilized HDAC6 decreased RPS5 expression to promote LF progression. The discovery of the ELAVL1/HDAC6/RPS5 axis may provide a new direction for understanding the pathogenesis of LF.
MATERIAL AND METHODS
Samples
A total of 33 patients with LC and 29 age-matched normal healthy individuals (undergoing health examinations) were recruited from the Hospital. The venous blood of each subject was collected and centrifuged to obtain the serum samples. Each subject signed a written informed consent form. Our study was approved by the Ethics Committee of Gansu Second People’s Hospital Northwest University Affiliated Hospital for Nationalities Hospital and performed in compliance with the Declaration of Helsinki.[17]
Cell culture, treatment, and transfection
Human HSC (LX-2; CL-0560, Procell, Wuhan, China), identified by short tandem repeat (STR) and determined by mycoplasma testing, was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (SH30022.01B, HyClone, Logan, UT, USA) at 37°C with 5% CO2. The LF cell model was established by treating LX-2 cells with 10 ng/mL TGF-β1 (AF-100-21C, PeproTech, Rocky Hill, NJ, USA) for 24 h, as previously described.[18] For transfection, LX-2 cells were transfected with siRNA against HDAC6/RPS5/ELAVL1 (si-HDAC6: F 5’-UAACGAUUCACUUA ACUCCAC-3’, R 5’-GGAGUUAAGUGAAUCGUUAAG-3’; si-RPS5: F 5’-UCUGUCACUCGGCGUAGACCA-3’, R 5’-GUCUACGCCGAGUGACAGAGA-3’; si-ELAVL1: F 5’-UCAUAACCAUUAGACAUUGUA-3’, R 5’-CAAUG UCUAAUGGUUAUGAAG-3’), polyclonal DNA (pcDNA) HDAC6 overexpression vector, and their controls using Lipofectamine 3,000 (L3000075, Invitrogen, Carlsbad, CA, USA) for 24 h before TGF-β1 treatment. The pcDNA HDAC6 overexpression vector was structured by inserting the polymerase chain reaction (PCR) products of HDAC6 (GenBank™ NM_001321225.2) into pcDNA3.1 vector (V79020, Invitrogen).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated by TRIzol reagent (15596026CN, Invitrogen), and cDNA was synthesized by Reverse Transcription Kit (RR716, Takara, Dalian, China). SYBR Green (RR820A, Takara) was mixed with cDNA and specific primers [Table 1] for PCR amplification following these thermocycling conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s, 55°C for 30 s, and 72°C for 30 s. Relative HDAC6 and RPS5 mRNA expressions were calculated by the 2−ΔΔCt method.
| Name | Primers for PCR (5’3’) |
|---|---|
| HDAC6 | |
| Forward | TCGCGGGGAAAAGGTCG |
| Reverse | GGGGGTTCTGCCTACTTCTT |
| RPS5 | |
| Forward | GAGTGACAGAGACGCTCAGG |
| Reverse | GCTCCACAATGGGACACTGA |
| GAPDH | |
| Forward | AGAAGGCTGGGGCTCATTTG |
| Reverse | AGGGGCCATCCACAGTCTTC |
HDAC6: Histone deacetylase 6, RPS5: Ribosomal protein S5, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, qRT-PCR: Quantitative real-time polymerase chain reaction, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
Western blot (WB)
Total protein was extracted by RIPA buffer (P0013B, Beyotime, Shanghai, China), quantified by BCA Kit (P0011, Beyotime), loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, and then transferred to polyvinylidene fluoride (PVDF) membranes (FFP24, Beyotime). After being blocked with non-fat milk and incubated with antibodies, the resulting membrane was treated with enhanced chemiluminescence (ECL) reagent (P0018AS, Beyotime) to visualize protein band using ImageQuant LAS 4000 Chemiluminescence imaging analyzer (General Electric Healthcare, Little Chalfont, Buckinghamshire, UK). Grayscale value was analyzed by ImageJ software (version 1.8.0, NIH, Bethesda, MD, USA), and relative protein expression was normalized by Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The antibodies (Abcam, Cambridge, CA, USA) included anti-HDAC6 (1:1000, ab1440), anti-α-smooth muscle actin α-SMA) (1:1000, ab5694), anti-collagen type 1 alpha 1 (COL1A1) (1:1000, ab34710), anti-RPS5 (1:2000, ab168823), anti-ELAVL1 (1:1000, ab200342), anti-GAPDH (1:2500, ab9485), and Goat anti-rabbit immunoglobulin G (IgG) (1:50000, ab205718).
Cell counting kit 8 (CCK8) assay
LX2 cells were seeded in 96-well plates and cultured with CCK8 reagent (C0041, Beyotime) after 48 h. Cell viability was assessed using a microplate reader (SpectraMax i3x, Molecular Devices, LLC, Sunnyvale, CA, USA) at 450 nm.
5-ethynyl-2’-deoxyuridine (EdU) assay
LX2 cells were stained with EdU solution (red fluorescence) and 4’,6-diamidino-2-phenylindole (DAPI) (blue fluorescence; C1002, Beyotime) using EdU Cell Proliferation Kit (C-10310-1, RiboBio, Guangzhou, China). Cell images were observed, and EdU-positive cells were analyzed under a fluorescence microscope (BX53, Olympus, Tokyo, Japan) with ImageJ software (version 1.8.0, NIH).
Transwell assay
LX2 cells suspended in serum-free medium were seeded in the upper part of the Matrigel-coated Transwell chambers (354480, Corning, Inc., Corning, NY, USA). The completed medium in the lower chamber was used as an inducer. After 24 h of incubation, invaded cells were fixed with polyformaldehyde, stained with crystal violet solution, and then counted under a microscope (IX71, Olympus).
Enzyme-linked immunosorbent assay (ELISA)
The culture supernatants of LX2 cells were harvested for detecting interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) levels using commercial Human IL-6 ELISA Kit (PI330, Beyotime) and Human TNF-α ELISA Kit (PT518, Beyotime), respectively, in accordance with the manufacturer’s instructions. Absorbance was analyzed by a microplate reader (SpectraMax i3x, Molecular Devices).
Bioinformatics prediction
Through the BioGRID database (https://thebiogrid.org/) prediction, we found that HDAC6 could interact with RPS5 [Supplementary Figure 1a]. The Encyclopedia of RNA Interactomes (ENCORI) website (https://rnasysu.com/encori/rbpClipRNA.php?source=mRNA) predicted that RNA binding protein ELAVL1 interacted with HDAC6 [Supplementary Figure 1b].
Co-immunoprecipitation (Co-IP) assay
LX-2 cells transfected with si-NC/si-HDAC6 were lysed, after which cell lysates were incubated with anti-RPS5 and Protein A/G beads (P2179S, Beyotime). The bead-bound complexes were collected and used for WB to detect the acetylation of RPS5 using anti-acetyl-lysine (Ac-lysine; 1:1000, ab190479, Abcam).
RNA immunoprecipitation (RIP) assay
LX-2 cells were lysed, and the collected supernatants were incubated with protein G-Sepharose beads (6511-100, Biovision, Milpitas, CA, USA) pre-coated with anti-ELAVL1 or anti-IgG. Immunoprecipitated RNA was isolated for testing HDAC6 enrichment using qRT-PCR.
mRNA stability assay
LX-2 cells transfected with si-NC/si-ELAVL1 were treated with 2 μg/mL Actinomycin D (Act D; A8030-1, Solarbio, Beijing, China) for 0, 3, 6, and 9 h. Then, total RNAs were extracted to examine HDAC6 mRNA expression by qRTPCR.
Statistical analysis
All experiments were performed in triplicate. Before conducting statistical analysis, we performed the Shapiro-Wilk normality test to confirm that all data were normally distributed. Data were presented as means ± SD and analyzed using GraphPad Prism 8.0 software (GraphPad, La Jolla, CA, USA). The differences between groups were evaluated by Student’s t-test (for 2 groups) or one-way analysis of variance, followed by Bonferroni post hoc test (for multiple groups). P < 0.05 was considered significant.
RESULTS
HDAC6 was upregulated in LC patients and TGF-β1-induced LF cell models
First, we measured HDAC6 expression in LC patients and normal healthy individuals. Through qRT-PCR, the results showed highly expressed HDAC6 in the serum of LC patients [P < 0.05, Figure 1a]. Furthermore, receiver operating characteristic curve analysis showed an HDAC6 AUC value of 0.9206 [P < 0.05, Figure 1b], suggesting that the population of LC patients and normal healthy individuals could be distinguished based on HDAC6 expression. In TGF-β1-induced LX-2 cells, we also detected a high HDAC6 expression [P < 0.05, Figure 1c and d].

- HDAC6 expression in LC patients and TGF-β1-induced LF cell models. (a) HDAC6 mRNA expressions in the serum of LC patients (n = 33) and normal healthy individuals (n = 29) were analyzed by qRT-PCR. (b) Receiver operating characteristic curve was used to analyze the value of HDAC6 expression in the diagnosis of LC. (c and d) HDAC6 mRNA and protein levels were determined by qRT-PCR and WB in LX-2 cells treated with or without TGF-β1 (n = 3). HDAC6: Histone deacetylase 6, LC: Liver cirrhosis, LF: Liver fibrosis, TGF-β1: Transforming growth factor β1, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot. ✶P < 0.05.
Silencing of HDAC6 alleviated TGF-β1-induced LX-2 cell proliferation, invasion, fibrosis, and inflammation
Loss of function experiments were performed to further determine the role of HDAC6 in LF progression. HDAC6 mRNA and protein expressions in LX-2 cells were reduced by the transfection of si-HDAC6 [P < 0.05, Figure 2a and b]. TGF-β1 treatment promoted LX-2 cell viability, EdU positive cells, and invaded cell numbers, while these effects were reversed by HDAC6 knockdown [P < 0.05, Figure 2c-f]. Moreover, TGF-β1 treatment increased the protein expression of fibrosis-related markers (α-SMA and COL1A1) and the levels of inflammatory factors (IL-6 and TNF-α) in LX-2 cells, while silencing HDAC6 abolished the promoting effect of TGF-β1 on the levels ofα-SMA, COL1A1, IL-6, and TNF-α [P < 0.05, Figure 2g-j]. Based on the results, we believe that HDAC6 could promote HSC activation to accelerate LF progression.
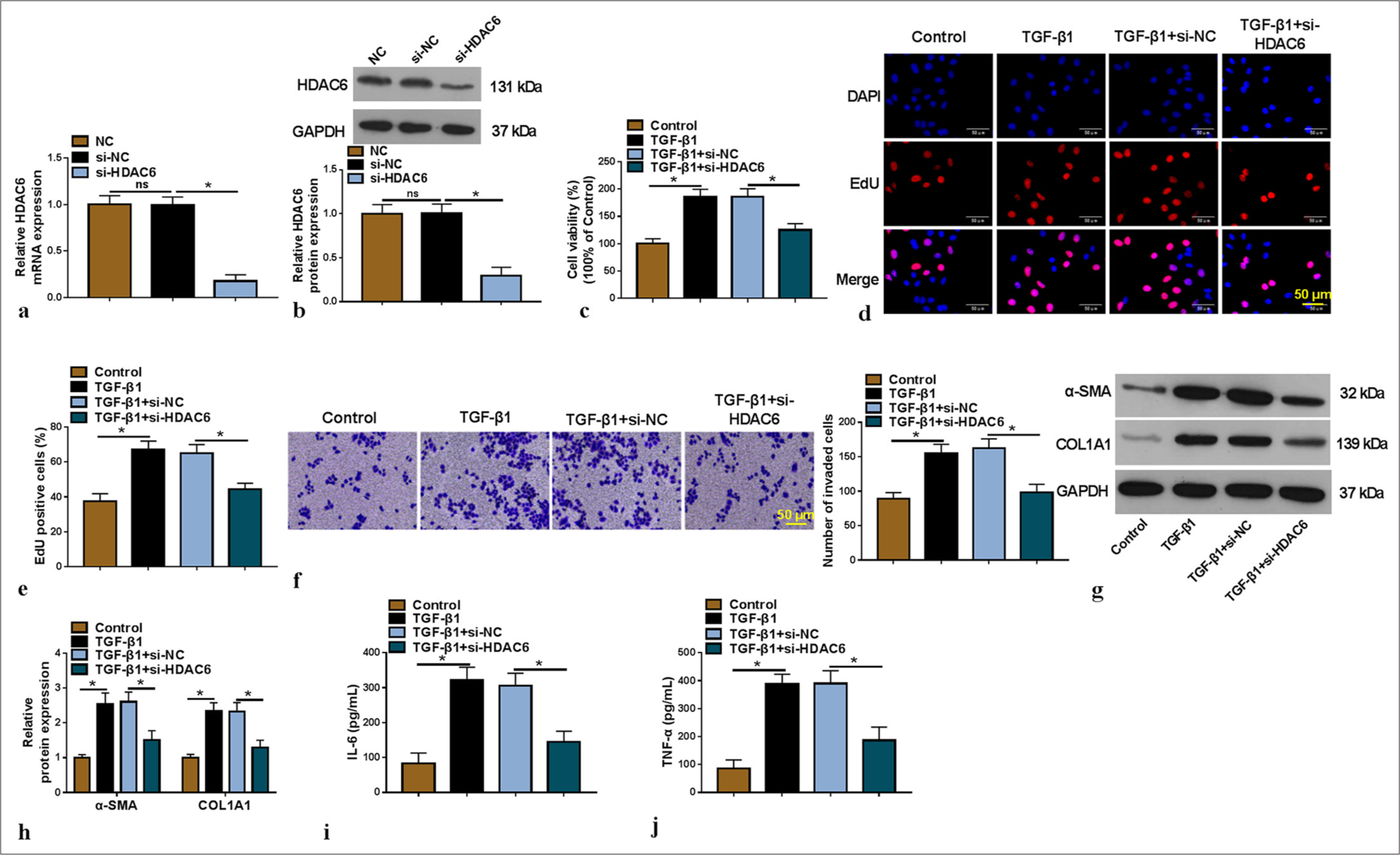
- Effect of si-HDAC6 on TGF-β1-induced HSC activation. (a and b) The transfection efficiency of si-HDAC6 was verified by qRTPCR and WB (n = 3). (c-j) TGF-β1-induced LX-2 cells were transfected with si-NC/si-HDAC6 (n = 3). CCK8 assay (c), EdU assay (×200) (d and e) and Transwell assay (f) were used to analyze cell proliferation and invasion. (g and h) The α-SMA and COL1A1 protein levels were examined by WB. (i and j) interleukin-6 and tumor necrosis factor alpha levels were determined by enzyme-linked immunosorbent assay. HDAC6: Histone deacetylase 6, TGF-β1: Transforming growth factor β1, HSC: Hepatic stellate cell, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot, CCK8: Cell counting kit 8, EdU: 5-ethynyl-2’-deoxyuridine. ✶P < 0.05.
HDAC6 inhibited the acetylation of RPS5
RPS5 protein levels were decreased in TGF-β1-induced LX-2 cells, while HDAC6 knockdown enhanced RPS5 expression [P < 0.05, Figure 3a]. Furthermore, RPS5 had a lower expression in the serum of LC patients [P < 0.05, Figure 3b], and its expression was negatively correlated with HDAC6 expression (r = −0.6478, P < 0.05, Figure 3c]. To further confirm the regulation of HDAC6 on the acetylation of RPS5, we performed a Co-IP assay. As shown in [Figure 3d], HDAC6 knockdown increased the acetylation levels of RPS5. Therefore, our data suggest that HDAC6 reduces RPS5 expression by inhibiting its acetylation.
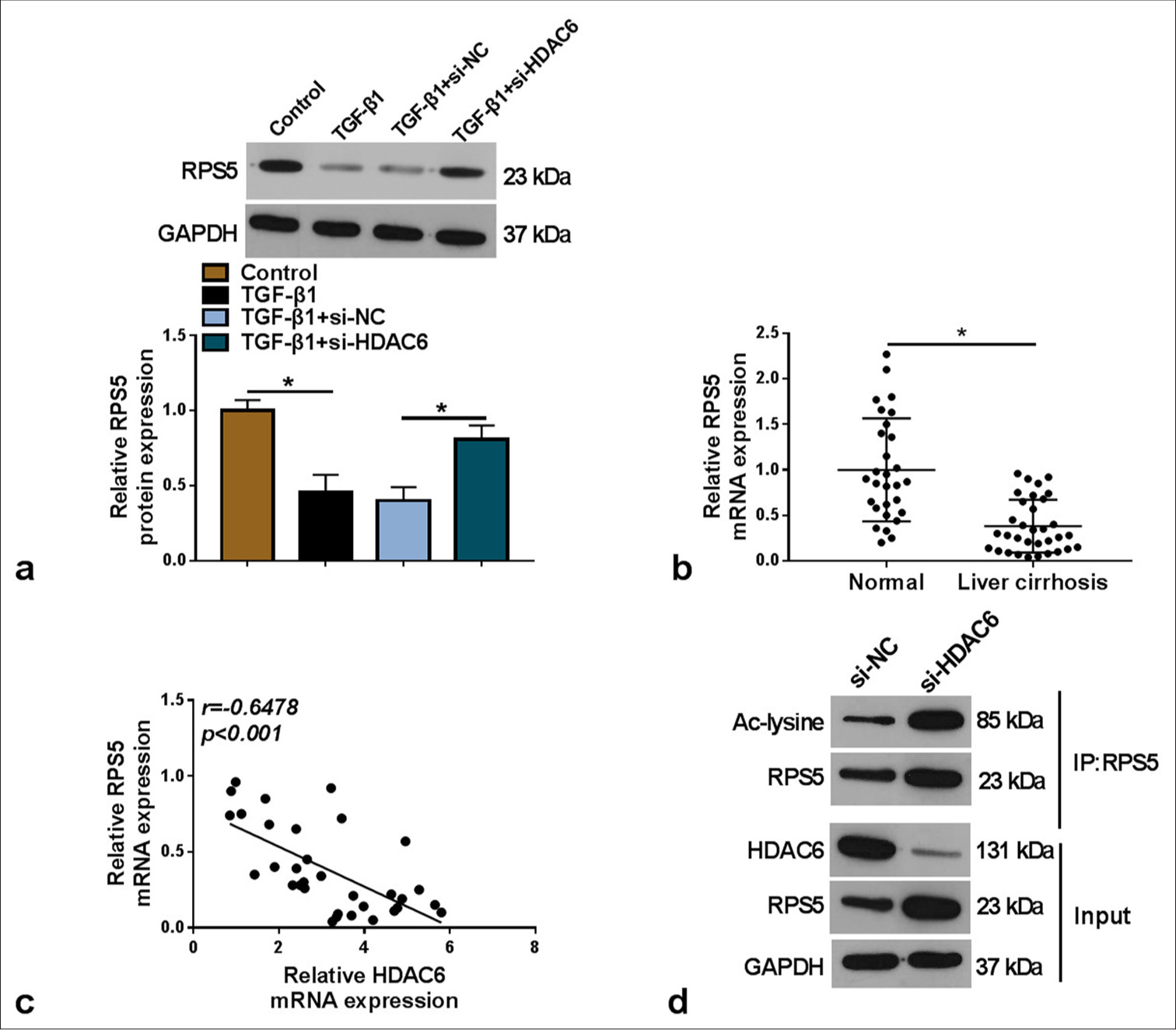
- HDAC6 regulated the acetylation of RPS5. (a) RPS5 protein expression was tested by WB in TGF-β1-induced LX-2 cells transfected with si-NC/si-HDAC6 (n = 3). (b) RPS5 mRNA expression was analyzed by qRT-PCR in the serum of LC patients (n = 33) and normal healthy individuals (n = 29). (c) Pearson correlation analysis was conducted to assess the interaction of HDAC6 and RPS5 expression. (d) Co-IP assay was performed to measure the regulation of si-HDAC6 on the acetylation of RPS5. HDAC6: Histone deacetylase 6, TGF-β1: Transforming growth factor β1, RPS5: Ribosomal protein S5, LC: Liver cirrhosis, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot, co-IP: Co-immunoprecipitation. ✶P < 0.05.
HDAC6 regulated RPS5 expression to affect TGF-β1-induced HSC activation
We performed rescue experiments to confirm further whether HDAC6 regulated LF progression by mediating RPS5 expression. The transfection of si-RPS5 was used to decrease RPS5 expression in LX-2 cells [P < 0.05, Figure 4a and b]. Then, TGF-β1-induced LX-2 cells were co-transfected with si-HDAC6 and si-RPS5. The inhibitory effect of si-HDAC6 on TGF-β1-induced LX-2 cell viability, EdU-positive cells, and invaded cell numbers could be overturned by RPS5 knockdown [P < 0.05, Figure 4c-f]. Furthermore, si-HDAC6-mediated the repressing effect on the levels of α-SMA, COL1A1, and IL-6, and TNF-α in TGF-β1-induced LX-2 cells were also reversed by RPS5 silencing [P < 0.05, Figure 4g-i].
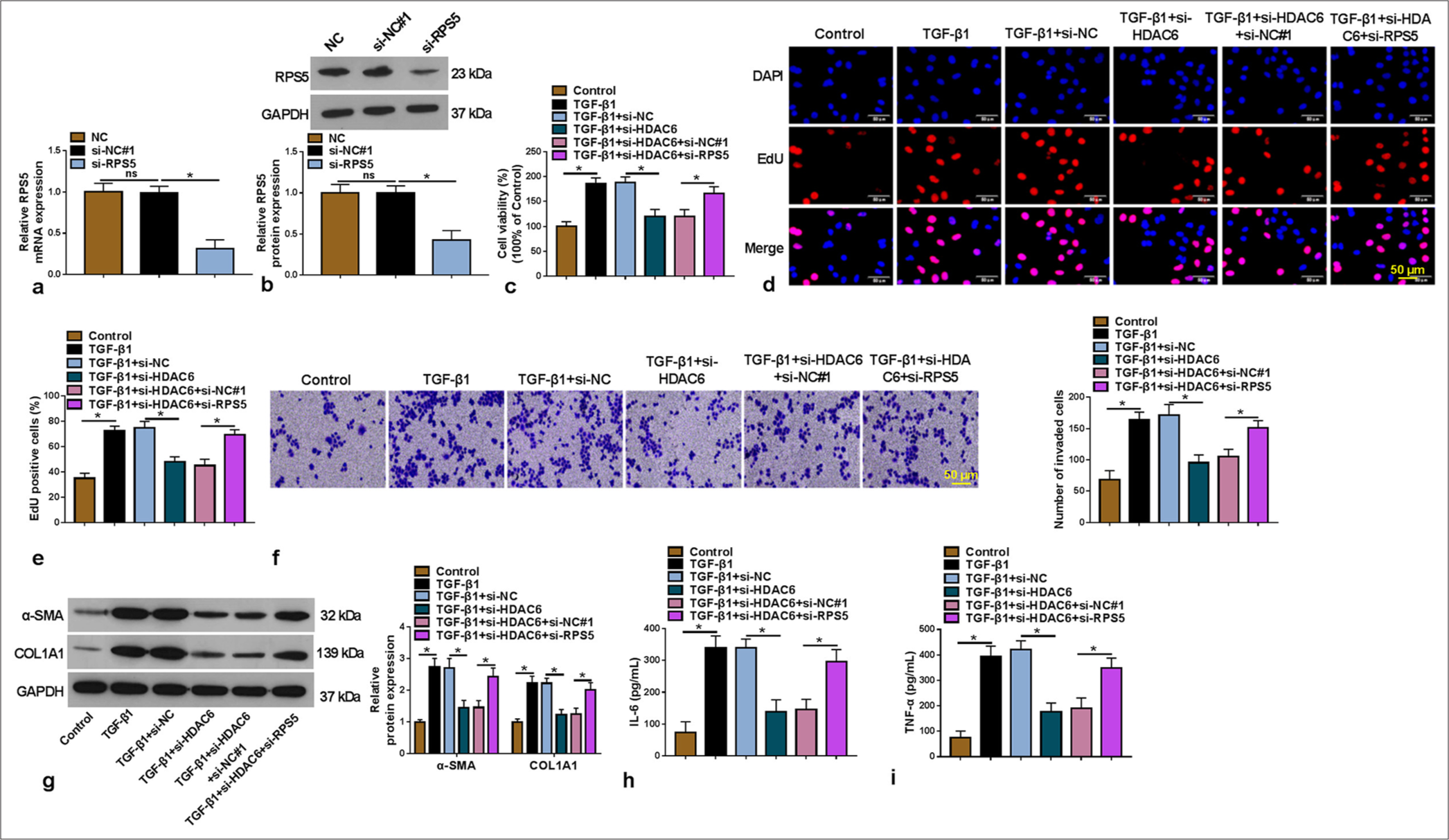
- Effects of si-HDAC6 and si-RPS5 on TGF-β1-induced HSC activation. (a and b) The transfection efficiency of si-RPS5 was detected using qRT-PCR and WB (n = 3). (c-i) TGF-β1-induced LX-2 cells were transfected with si-NC/si-HDAC6/si-NC#1/si-RPS5 (n = 3). Cell proliferation and invasion were measured using CCK8 assay (c), EdU assay (200×) (d and e) and Transwell assay (f). (g) WB was used to detect the α-SMA and COL1A1 protein levels. (h and i) enzyme-linked immunosorbent assay was performed to examine the interleukin-6 and tumor necrosis factor alpha levels. HDAC6: Histone deacetylase 6, TGF-β1: Transforming growth factor β1, HSC: Hepatic stellate cell, RPS5: Ribosomal protein S5, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot, CCK8: Cell counting kit 8, EdU: 5-ethynyl-2’-deoxyuridine. ✶P < 0.05.
ELAVL1-stabilized HDAC6 mRNA expression
RIP assay results indicated that HDAC6 enrichment was increased by anti-ELAVL1 [P < 0.05, Figure 5a], confirming the interaction between them. Furthermore, the transfection of si-ELAVL1 was used to reduce ELAVL1 mRNA and protein expression [P < 0.05, Figure 5b and c]. After Act D treatment, ELAVL1 knockdown inhibited the mRNA stability of HDAC6 [P < 0.05, Figure 5d]. We also discovered that ELAVL1 knockdown could decrease HDAC6 mRNA and protein levels in TGF-β1-induced LX-2 cells [P < 0.05, Figure 5e and f]. These data suggest that ELAVL1 might increase HDAC6 expression by stabilizing its mRNA.
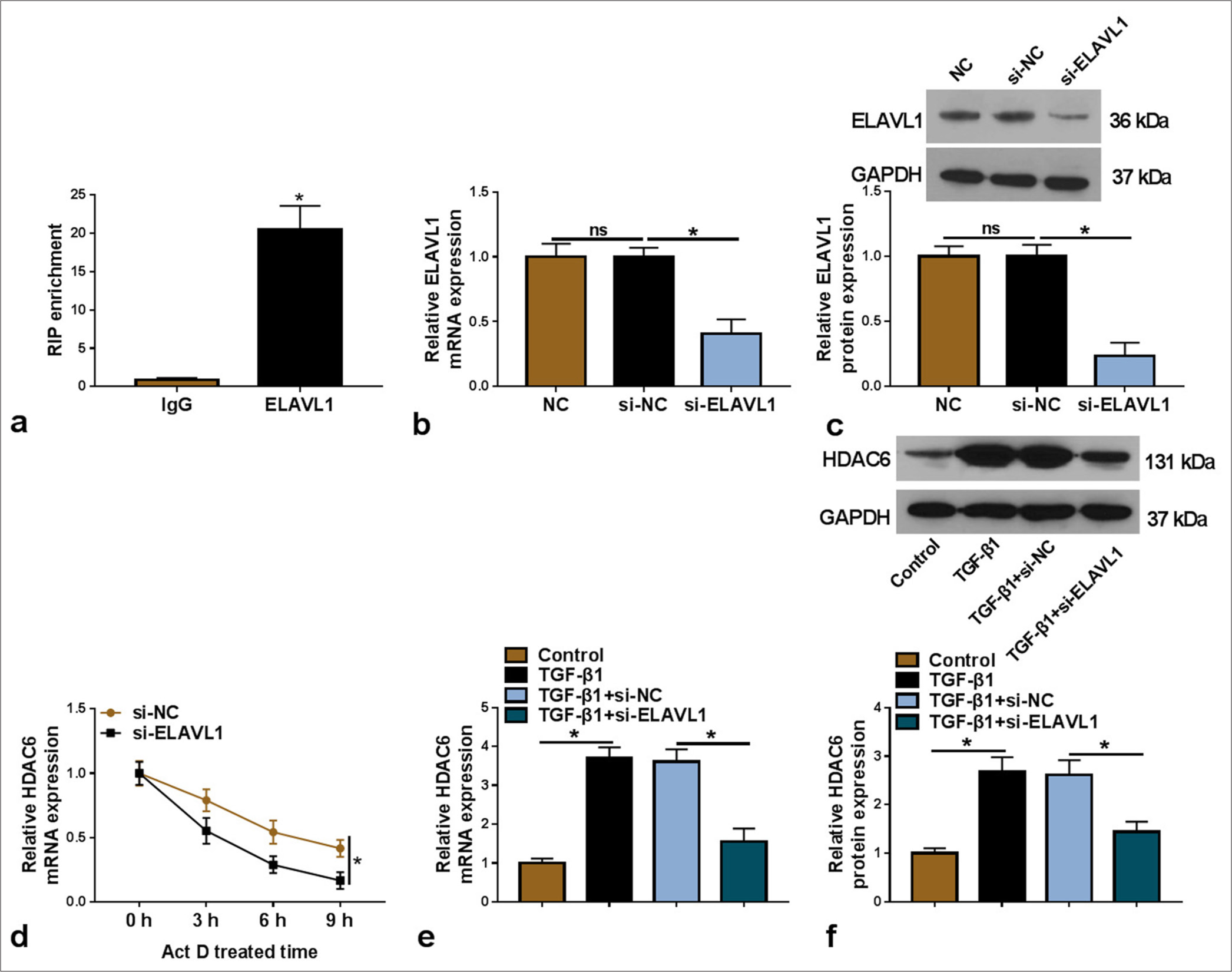
- ELAVL1 regulated HDAC6 mRNA expression. (a) RIP assay was used to confirm the interaction between ELAVL1 and HDAC6 (n = 3). (b and c) The transfection efficiency of si-ELAVL1 was verified by qRT-PCR and WB (n = 3). (d) Act D assay was used to confirm the regulation of si-ELAVL1 on HDAC6 mRNA stability (n = 3). (e and f) HDAC6 mRNA and protein levels in TGF-β1-induced LX-2 cells transfected with si-NC/si-ELAVL1 were detected by qRT-PCR and WB (n = 3). HDAC6: Histone deacetylase 6, TGF-β1: Transforming growth factor β1, HSC: Hepatic stellate cell, ELAVL1: Embryonic lethal abnormal vision like 1, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot, RIP: RNA immunoprecipitation, Act D: Actinomycin D. ✶P < 0.05.
ELAVL1 negatively regulated RPS5 expression by stabilizing HDAC6
We conducted further analysis to verify further the regulation of ELAVL1 and HDAC6 on RPS5 expression. HDAC6 overexpression vector was transfected into LX-2 cells to enhance HDAC6 expression [P < 0.05, Figure 6a and b]. In TGF-β1-induced LX-2 cells, ELAVL1 knockdown markedly increased RPS5 mRNA and protein levels, and this effect could be abolished by HDAC6 overexpression [P < 0.05, Figure 6c and d]. Therefore, these results indicate that ELAVL1 stabilized HDAC6 to reduce RPS5 expression.
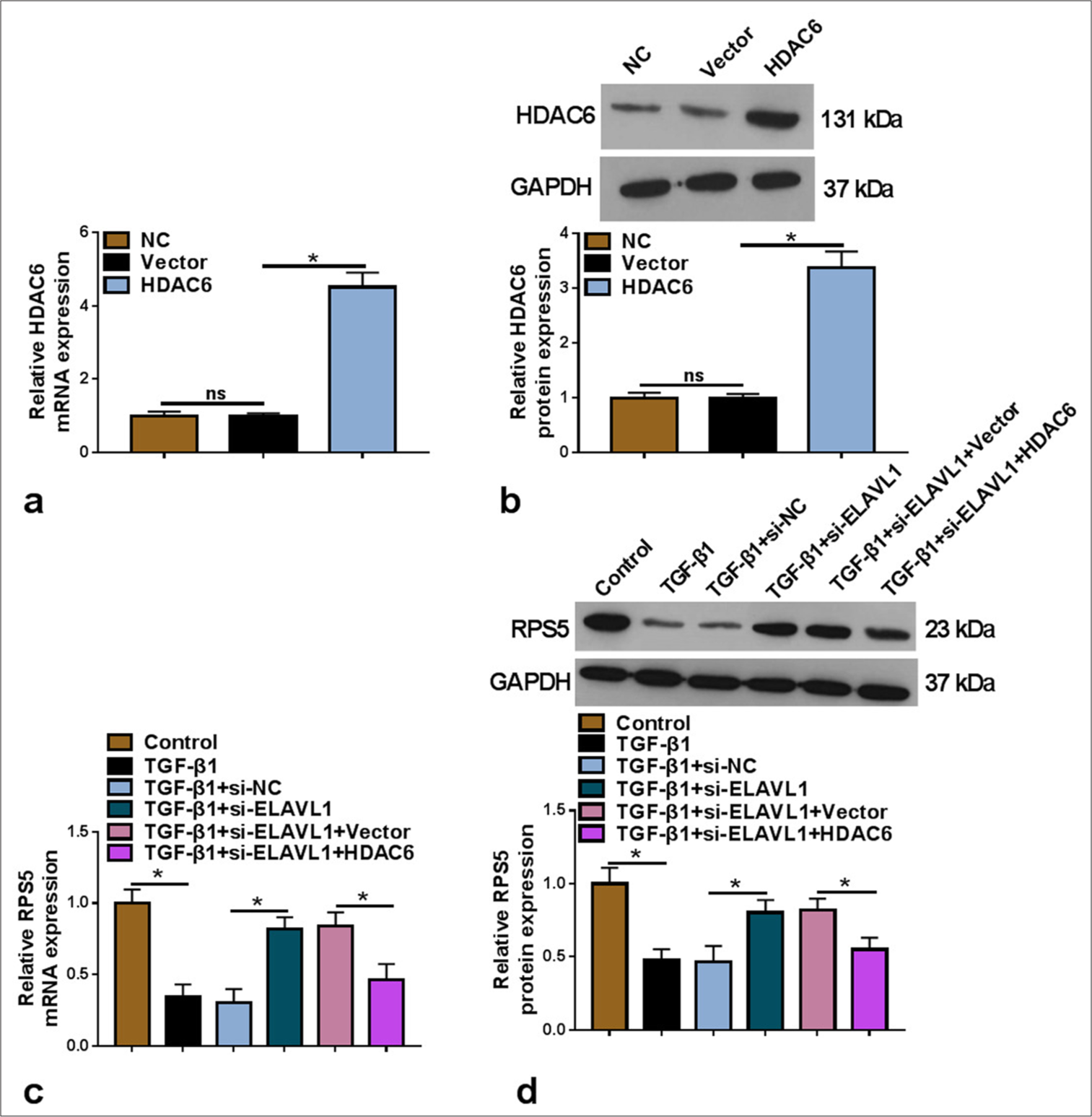
- Effects of si-ELAVL1 and HDAC6 on RPS5 expression. (a and b) The transfection efficiency of HDAC6 overexpression vector was confirmed by qRT-PCR and WB (n = 3). (c and d) RPS5 mRNA and protein levels were examined by qRT-PCR and WB in TGF-β1-induced LX-2 cells transfected with si-NC/si-ELAVL1/Vector/HDAC6 (n = 3). HDAC6: Histone deacetylase 6, TGF-β1: Transforming growth factor β1, ELAVL1: Embryonic lethal abnormal vision like 1, RPS5: Ribosomal protein S5, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot. ✶P < 0.05.
DISCUSSION
LF is a major global health challenge and a significant risk factor for liver diseases, including LC and HCC.[19,20] HSC activation is the main driving force of hepatic fiber formation.[7,21] The inducers of HSC activation include TGF-β, PDGF-β, and inflammatory cytokines.[22] Studies have found that the remission of LF is related to the inactivation of HSC;[23] thus, inhibiting HSC activation has become a promising direction for LF treatment.[24] In this study, we identified a new mechanism that regulated HSC activation, thus providing insights into the mitigation of LF progression.
HDAC6 has been shown to play an important role in liver-related diseases. In particular, previous studies revealed that sulforaphane inhibited cell ferroptosis to alleviate acute liver failure by decreasing HDAC6 expression.[25] A study reported that HDAC6 had elevated expression in HCC tissues and that its upregulation promoted HCC cell proliferation.[26] Dai et al. found that the upregulation of HDAC6 might be associated with the poor prognosis and reduced survival rate of liver cancer patients, as it could facilitate cancer cell proliferation and invasion.[27]
Furthermore, the role of HDAC6 and its inhibitors in the occurrence and progression of LF has been well studied.[28] A previous study confirmed the high HDAC6 expression in fibrotic rats and that decreased HDAC6 expression might relieve the LF process.[11] Consistent with such findings, we detected the upregulated HDAC6 in LC patients and TGF-β1-induced LF cell models. Loss of functional assay showed that the downregulation of HDAC6 mitigated TGF-β1-induced LX-2 cell proliferation, invasion, fibrosis, and inflammation, confirming that HDAC6 might contribute to HSC activation to aggravate LF progression.
RPS5 is one of the major proteins that bind to RNA.[12] It had been revealed that RPS5 expression was reduced during fibrotic stimulation and that matrine-stabilized RPS5 prevented myofibroblast generation and the development of cardiac fibrosis.[29] Importantly, Xu et al. suggested that RPS5 suppressed HSC activation to alleviate the DMN/BDL-induced LF process.[15] In the present study, we found that RPS5 expression decreased in LC patients and that HDAC6 downregulated RPS5 expression by repressing its acetylation. Furthermore, the repressing effect of si-HDAC6 on TGF-β1-induced HSC proliferation, invasion, fibrosis, and inflammation could be abolished by RPS5 knockdown, indicating that HDAC6 facilitated the LF process by decreasing RPS5 acetylation.
ELAVL1 mainly binds to cellular mRNA to regulate its stability. ELAVL1 has also been reported to inhibit CD4-positive T-cell invasion in prostate cancer by promoting PD-L1 mRNA stability.[30] Furthermore, a study reported that CircRHOBTB3 interacted with ELAVL1 to reduce PTBP1 mRNA stability, thus inhibiting colorectal cancer metastasis.[31] It has also been suggested that ELAVL1 was upregulated in mouse fibrotic liver and that its expression was positively correlated with the levels of fibrosis-related proteins.[16] Additional studies further revealed that the silencing of ELAVL1 attenuated fibrosis development and HSC activation.[32] These data confirm the active role of ELAVL1 in the LF process. In the present study, we found that ELAVL1 stabilized HDAC6 mRNA stability to promote its expression. Furthermore, our results revealed that ELAVL1 could decrease RPS5 expression by stabilizing HDAC6. These data refined our hypothesis on the ELAVL1/HDAC6/RPS5 axis.
Certainly, there are some limitations of this study. At present, we use only one type of HSC for exploration, which is not sufficient. In general, two cell lines are selected for disease mechanism research, and the use of two or more cell lines can help researchers understand the complexity of the disease more fully. In the future, we will consider repeating the present study using other HSCs to confirm further the important function of the ELAVL1/HDAC6/RPS5 axis in LF progression. At present, we only examined the expression of HDAC6 in the serum of LC patients but did not analyze its correlation with the clinical characteristics of patients.
In the future, it is necessary to collect patient information further, provide more data on the expression of HDAC6 in different stages of LF, and analyze its correlation with disease progression. Thus far, we have only revealed that ELAVL1 acts as an RNA-binding protein to stabilize HDAC6 expression but have yet to perform the corresponding functional experiments. In the future, rescue experiments are still needed to clarify the regulation of ELAVL1 on the HDAC6/RPS5 axis and its effect on HSC activation. Doing so can provide more evidence for how exactly the ELAVL1/HDAC6/RPS5 axis can mediate LF progression.
SUMMARY
Our study revealed that ELAVL1-stabilized HDAC6 facilitated HSC activation to promote LF progression through the inhibition of RPS5 acetylation modification. The proposed ELAVL1/HDAC6/RPS5 axis may play a key role in LF progression, thus providing a new theoretical basis for the treatment of LC.
ACKNOWLEDGMENT
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
All data and materials are available from the corresponding author with reasonable request.
ABBREVIATIONS
CCK8: Cell counting kit 8
co-IP: Co-immunoprecipitation
EdU: 5-ethynyl-2’-deoxyuridine
ELAVL1: Embryonic lethal abnormal vision like 1
HCC: Hepatocellular carcinoma
HDAC6: Histone deacetylase 6
HDACs: Histone deacetylases
HSC: Hepatic stellate cell
LC: Liver cirrhosis
LF: Liver fibrosis
qRT-PCR: Quantitative real-time polymerase chain reaction
RIP: RNA immunoprecipitation
RPS5: Ribosomal protein S5
TGF-β1: Transforming growth factor β1
WB: Western blot
AUTHOR CONTRIBUTIONS
QW: Conception and design; WJZ: Administrative support; JPW: Provision of study materials or patients; LZ: Collection and assembly of data; YWQ and YC: Data analysis and interpretation; QW: Funding acquisition. All authors contributed to manuscript writing. All authors gave final approval of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Our study was approved by the Ethics committee of Gansu Second People’s Hospital Northwest University Affiliated Hospital for Nationalities Hospital (XBMZ-YX-20230266). And each subject signed a written informed consent.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: This research is supported by Basic scientific research funds for central universities in 2020 (31920200110).
References
- Burden of liver diseases in the world. J Hepatol. 2019;70:151-71.
- [CrossRef] [PubMed] [Google Scholar]
- From liver to hormones: The endocrine consequences of cirrhosis. World J Gastroenterol. 2024;30:1073-95.
- [CrossRef] [PubMed] [Google Scholar]
- Human umbilical cord-derived mesenchymal stem cells attenuate hepatic stellate cells activation and liver fibrosis. Mol Biol Rep. 2024;51:734.
- [CrossRef] [PubMed] [Google Scholar]
- Author correction: Mesenchymal stromal cells in hepatic fibrosis/cirrhosis: From pathogenesis to treatment. Cell Mol Immunol. 2023;20:687-8.
- [CrossRef] [Google Scholar]
- Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives. Mech Ageing Dev. 2021;199:111572.
- [CrossRef] [PubMed] [Google Scholar]
- The role of histone acetylation modification in dental tissue-derived mesenchymal stem cells and odontogenesis. Cell Reprogram. 2023;25:11-9.
- [CrossRef] [PubMed] [Google Scholar]
- HDAC6 as privileged target in drug discovery: A perspective. Pharmacol Res. 2021;163:105274.
- [CrossRef] [PubMed] [Google Scholar]
- A novel HDAC6 inhibitor attenuate APAP-induced liver injury by regulating MDH1-mediated oxidative stress. Int Immunopharmacol. 2024;131:111861.
- [CrossRef] [PubMed] [Google Scholar]
- Histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates liver fibrosis by suppressing the transforming growth factor-β1 signal pathway. Hepatobiliary Pancreat Dis Int. 2018;17:423-9.
- [CrossRef] [PubMed] [Google Scholar]
- Eukaryotic ribosomal protein S5 of the 40S subunit: Structure and function. Int J Mol Sci. 2023;24:3386.
- [CrossRef] [PubMed] [Google Scholar]
- Matrine alleviates hypoxia-induced inflammation and pulmonary vascular remodelling via RPS5/NF-κB signalling pathway. J Biochem Mol Toxicol. 2024;38:e23583.
- [CrossRef] [PubMed] [Google Scholar]
- Unraveling the role of RNA-binding proteins, with a focus on RPS5, in the malignant progression of hepatocellular carcinoma. Int J Mol Sci. 2024;25:773.
- [CrossRef] [PubMed] [Google Scholar]
- Bioactive compound reveals a novel function for ribosomal protein S5 in hepatic stellate cell activation and hepatic fibrosis. Hepatology. 2014;60:648-60.
- [CrossRef] [PubMed] [Google Scholar]
- Essential roles of RNA-binding protein HuR in activation of hepatic stellate cells induced by transforming growth factor-β1. Sci Rep. 2016;6:22141.
- [CrossRef] [PubMed] [Google Scholar]
- The 2024 revision of the Declaration of Helsinki: a modern ethical framework for medical research. Postgrad Med J 2024:qgae181.
- [CrossRef] [PubMed] [Google Scholar]
- FAT10 Silencing prevents liver fibrosis through regulating SIRT1 expression in hepatic stellate cells. Int J Med Sci. 2023;20:557-65.
- [CrossRef] [PubMed] [Google Scholar]
- The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int J Mol Sci. 2019;20:1723.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology. 2020;158:1728-44.e14.
- [CrossRef] [PubMed] [Google Scholar]
- Docosahexaenoic acid attenuates carbon tetrachloride-induced hepatic fibrosis in rats. Int Immunopharmacol. 2017;53:56-62.
- [CrossRef] [PubMed] [Google Scholar]
- Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073-83.e22.
- [CrossRef] [PubMed] [Google Scholar]
- GATA4 induces liver fibrosis regression by deactivating hepatic stellate cells. JCI Insight. 2021;6:e150059.
- [CrossRef] [PubMed] [Google Scholar]
- Sulforaphane, an NRF2 agonist, alleviates ferroptosis in acute liver failure by regulating HDAC6 activity. J Integr Med. 2023;21:464-73.
- [CrossRef] [PubMed] [Google Scholar]
- HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013;587:880-6.
- [CrossRef] [PubMed] [Google Scholar]
- HDAC6 promotes aggressive development of liver cancer by improving egfr mRNA stability. Neoplasia. 2023;35:100845.
- [CrossRef] [PubMed] [Google Scholar]
- Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie. 2015;116:61-9.
- [CrossRef] [PubMed] [Google Scholar]
- Matrine attenuates pathological cardiac fibrosis via RPS5/p38 in mice. Acta Pharmacol Sin. 2021;42:573-84.
- [CrossRef] [PubMed] [Google Scholar]
- ELAVL1 regulates PD-L1 mRNA stability to disrupt the infiltration of CD4-positive T cells in prostate cancer. Neoplasia. 2024;57:101049.
- [CrossRef] [PubMed] [Google Scholar]
- Circular RNA circRHOBTB3 represses metastasis by regulating the HuR-mediated mRNA stability of PTBP1 in colorectal cancer. Theranostics. 2021;11:7507-26.
- [CrossRef] [PubMed] [Google Scholar]
- Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology. 2012;56:1870-82.
- [CrossRef] [PubMed] [Google Scholar]




