


Translate this page into:
Emerging regulators of gastric cancer angiogenesis: Synergistic effects of regulator of G protein signaling 4 and midkine

*Corresponding author: HaiPing Song, Department of Medical Oncology, Qingdao Central Hospital, University of Health and Rehabilitation Sciences, Qingdao, China. 18561856653@163.com
-
Received: ,
Accepted: ,
How to cite this article: He Y, Li H, Li K, Song H. Emerging regulators of gastric cancer angiogenesis: Synergistic effects of regulator of G protein signaling 4 and midkine. CytoJournal. 2025;22:26. doi: 10.25259/Cytojournal_212_2024
Abstract
Objective
Globally, gastric cancer (GC) is among the most prevalent cancers. The development and spread of stomach cancer are significantly influenced by angiogenesis. However, the molecular mechanisms underlying this process remain unclear. This study aimed to investigate the role of the regulator of G protein signaling 4 (RGS4) in GC angiogenesis and its potential mechanisms.
Material and Methods
Through in vitro and in vivo experiments, including tube formation assays and xenograft models in nude mice, we evaluated the effects of RGS4 on GC angiogenesis and metastasis. In addition, we employed techniques such as immunoprecipitation and immunofluorescence double staining to explore the interaction between RGS4 and midkine (MDK). Survival analysis was also performed to evaluate the association between the prognosis of patients with GC and the expression levels of RGS4 and MDK.
Results
Our findings revealed that RGS4 is a crucial factor in GC metastasis, significantly inducing angiogenesis. Further studies indicated that RGS4 directly interacts with MDK and upregulates its expression. By upregulating MDK, RGS4 stimulates the angiogenesis and metastasis of GC. Furthermore, a poor prognosis for patients with GC is directly linked to high expression of RGS4 and MDK.
Conclusion
This work is the first to clarify the molecular mechanism by which RGS4 upregulates MDK expression to increase GC angiogenesis. These findings not only enhance our understanding of the mechanisms underlying GC progression but also provide potential targets for developing new anti-angiogenic and antimetastatic therapies. RGS4 and MDK could serve as effective biomarkers for predicting the prognosis of patients with GC and offer new insights into personalized treatment approaches.
Keywords
Angiogenesis
Midkine
Regulator of G protein signaling 4

INTRODUCTION
With a high incidence and mortality rate, gastric cancer (GC) is one of the most prevalent and lethal cancers in the world.[1-3] Despite recent advancements in the diagnosis and treatment of GC, its prognosis remains poor, primarily due to its propensity for invasion and metastasis.[4,5] The development of new blood vessels, or angiogenesis, is a critical component in tumor growth and metastasis.[6,7] Angiogenesis speeds up the growth and spread of tumors by boosting the flow of oxygen and nutrients.[8] Thus, understanding the molecular mechanisms of GC angiogenesis is vital for developing new therapeutic strategies.[9,10] However, the specific regulatory mechanisms of angiogenesis in GC remain unclear.[11]
Regulator of G protein signaling 4 (RGS4) is an important signaling molecule that primarily functions by modulating the signal transduction of G protein-coupled receptors.[12] RGS4 has been shown in earlier research to have a major regulatory role in a number of malignancies, such as glioblastoma, prostate cancer, and breast cancer.[12-14] For instance, RGS4 has been found to promote cell proliferation and migration in glioblastoma cells by regulating the nuclear factor kappa-B signaling pathway.[15] However, the precise function and processes of RGS4 in GC remain unknown.
Midkine (MDK) is a low-molecular-weight growth factor with multiple biological functions, including promoting cell proliferation, migration, and angiogenesis.[16,17] MDK is tightly linked to tumor invasion and metastasis and is significantly expressed in a variety of malignancies. For instance, MDK stimulates the Notch signaling system, which in turn promotes angiogenesis in hepatocellular cancer.[18,19] In non-small cell lung cancer, MDK promotes tumor angiogenesis by regulating the expression of vascular endothelial growth factor (VEGF).[20] However, the upstream regulatory mechanisms of MDK, particularly in GC, remain incompletely understood.
Despite studies revealing the significant roles of RGS4 and MDK in various cancers, research on RGS4 promoting GC angiogenesis through the upregulation of MDK expression is still lacking. Existing studies mainly focus on the individual functions and mechanisms of RGS4 and MDK, with a shortage of systematic research on their interactions. Furthermore, although RGS4 and MDK are both highly expressed in GC and closely linked to patient prognosis, their exact molecular mechanisms remain unclear. Therefore, investigating the interaction and regulatory mechanisms between RGS4 and MDK in GC angiogenesis is of significant scientific and clinical value.
This work aimed to investigate the function and molecular mechanisms of RGS4 in the angiogenesis of GC. We hypothesize that RGS4 promotes angiogenesis in GC by upregulating MDK expression. To validate this hypothesis, we analyzed the relationship between RGS4 and clinical pathological features. We then used in vitro and in vivo tests to assess the effect of RGS4 on the angiogenic potential of GC cells. Finally, we investigated the specific mechanisms by which RGS4 regulates MDK expression through molecular biology techniques. This study is the first to reveal the critical role of RGS4 in GC angiogenesis and elucidate its mechanism of action through the upregulation of MDK expression. We intend to improve the prognosis of patients with GC by developing advanced treatment plans by thoroughly understanding the roles and mechanisms of RGS4 and MDK in this disease.
MATERIAL AND METHODS
Cell line culture, grouping, plasmid transfection, and transfection efficiency detection
NCI-N87 cells (BFN60808579, ATCC, Manassas, VA, USA) were retrieved from liquid nitrogen storage and rapidly thawed in a 37°C℃ water bath. NCI-N87 cells were mycoplasma-free, and short tandem repeat analysis revealed that they were derived from its parental cells. After centrifugation, the cell pellets were resuspended in Roswell Park Memorial Institute 1640 (RPMI1640) medium (12633020, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (BL201A, Biosharp Life Science, Anhui, China) and 1% double antibody (penicillin-streptomycin mixture, BL505A, Biosharp Life Science, Anhui, China). The cells were then transferred to culture flasks and incubated in a sterile 37°C incubator with 5% carbon dioxide (CO2) to promote cell growth. Following their 70%–80% confluence, the cells were reseeded into the proper culture flasks or plates after being digested with 0.25% trypsin (15400054, Gibco, Life Technologies, Rockville, MD, USA). The cells were allowed to adhere for 12 h in an incubator, ensuring that they were firmly attached. All experimental cells were in the logarithmic growth phase. Logarithmic phase NCI-N87 cells were seeded into six-well plates (1.2 × 106 cells/well). The RGS4 overexpression plasmid and empty vector plasmid were diluted with 250 μL of Opti-MEM (31985070, Thermo Fisher Scientific, Waltham, MA, USA) once the cells had reached 40% confluence. Similarly, 5 μL of the transfection reagent Lipofectamine™ 2000 (11668500, Thermo Fisher Scientific, Waltham, MA, USA) was diluted with 250 μL of Opti-MEM and left to stand for 5 min. The gene sequence dilutions were then gently mixed with the Lipofectamine™ 2000 solution and left to form complexes for 20 min. The mixtures were added dropwise to the wells, and the medium in each well was topped up to 2 mL with Opti-MEM. The cells were grown for an extra 48 h before collection, and the medium was changed to a complete medium after 6 h of transfection. The expression of RGS4 protein in each group was determined using Western blot analysis to confirm gene expression rates.
Western blot
Total protein from each group of cells was extracted using cell lysis buffer (radioimmunoprecipitation assay: phenylmethanesulfonyl fluoride = 100:1, BL509A, Biosharp Life Science, Anhui, China). Protein quantification was performed using the bicinchoninic acid assay protein assay kit (BL524A, Biosharp Life Science, Anhui, China). Total protein was separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis. After electrophoresis, the proteins were transferred to a polyvinylidene fluoride membrane (IPVH00010, Millipore Corporation, Billerica, MA, USA) through a wet transfer method. The membrane was blocked with 5% skim milk at room temperature for 2 h and then incubated with primary antibody (1:1000, Abcam, Cambridge, MA, USA) overnight at 4°C. The next day, the membrane was washed 3 times with tris-buffered saline with tween-20 (TBST, 10 min each), incubated with secondary antibody (1:5000, SA00001-2, Proteintech, Hubei, China) for 1 h, and washed 3 times again with TBST (10 min each). Enhanced chemiluminescence chemiluminescent reagents (BL520b, Biosharp Life Science, Anhui, China) were applied, and the membrane was developed using a chemiluminescence gel imaging system (Amersham ImageQuant 800, Cytiva, Uppsala, Sweden). The band intensity was analyzed using ImageJ software (v1.3, National Institutes of Health, Bethesda, MD, USA), and the relative protein expression was determined by the ratio of gray values.
The primary antibodies were as follows: RGS4 (ab97307), VEGF-A (ab46154), VEGF-B (ab133606), VEGF-C (AB9546), MDK (ab52637), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab9485).
Immunohistochemistry
Paraffin-embedded tissue blocks were sectioned continuously, deparaffinized with xylene, and rehydrated. Antigen retrieval was performed with Ethylenediaminetetraacetic Acid (pH 9.0,60126ES02, Yeasen, Shanghai, China) for 20 min, followed by phosphate buffer saline (PBS, ST447, Beyotime Biotechnology, Shanghai, China) washes. After 10 min of blocking endogenous peroxidase activity with 3% H2O2 and then with 10% goat serum (C0265, Beyotime Biotechnology, Shanghai, China) for 30 min, we added 100 μL of anti-CD31 antibody (ab28364), RGS4 (ab97307), and MDK (ab52637). The mixture was then rinsed with PBS, and all primary antibodies were obtained from Abcam (Cambridge, MA, USA). The sections were incubated at 37°C for 60 min, washed with PBS, and added with 100 μL of goat anti-rabbit immunoglobulin G (IgG) polymer (ab6721, Abcam, Cambridge, MA, USA). After 30 min of incubation at room temperature and PBS washes, diaminobenzidine chromogen (P0202, Beyotime Biotechnology, Shanghai, China) was applied, followed by hematoxylin (H810910, Macklin, Shanghai, Beijing) counterstaining, xylene (X821391, MACKLIN, Shanghai, Beijing) clearing, and neutral resin (N861409, MACKLIN, Shanghai, Beijing) mounting. We used ImageJ and fluorescence microscopy (BX51FL, Olympus Corporation, Tokyo, Japan) for image analysis.
Tail vein injection of GC cells (NCI-N87) in nude mice
A total of 42 female nude mice, aged 6–8 weeks and weighing between 18 and 20 g, were purchased from the animal experiment center. The experimental mice were housed in a specific pathogen-free (SPF)-grade laboratory with controlled conditions: 24°C ± 2°C, relative humidity of 40– 60%, and a 12 h light/12 h dark cycle, with free access to food and water. After a 3-day acclimation period, the mice were fed standard mouse pellet feed throughout the experiment. The mice were restrained in a mouse holder, and their tails were warmed with a warm water-soaked cotton ball to dilate the tail veins. The tail was then disinfected with alcohol. Using a 1 mL syringe, 0.1 mL of the NCI–N87 cancer cell suspension (2 × 107/mL) was drawn and injected into the lateral tail vein. The needle was inserted approximately 0.3 cm into the tail vein. Upon withdrawal of the plunger, blood backflow into the syringe indicated proper placement within the vein. The cancer cell suspension was then rapidly injected over a period of 3 s. The tail was disinfected again, and the mice were observed for any abnormalities before being returned to SPF conditions for maintenance. After all the experiments, the mice were killed by neck dislocation. All animal procedures were performed in accordance with the guidelines for the care and use of Laboratory Animals in the Hospital. The study was approved by the Institutional Animal Care and Use Committee of the Hospital.
Hematoxylin and eosin (H&E) staining of lung tissues
After 6 weeks, the mice were euthanized, and their lung tissues were fixed in 10% neutral formalin (BL388A, Biosharp Life Science, Anhui, China) for 24 h. Following dehydration, the fixed tissues were embedded in paraffin and cut into slices that were 4 μm thick. The sections were stained with H&E (C0105M, Beyotime Biotechnology, Shanghai, China) and examined for pathological changes. The number of lung nodes was observed by microscopy (BX46, Olympus Corporation, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA) detection of VEGFA, VEGFB, and VEGFC levels
The cells to be tested were collected, and the supernatant was used to detect the levels of VEGFA, VEGFB, and VEGFC. The specific operational steps followed the instructions of the VEGFA (ab222510), VEGFB (ab277458), and VEGFC (ab100664) ELISA kits. In brief, as per the ELISA kit protocols (Abcam, Cambridge, MA, USA), the standards and protein samples were diluted, and 100 μL of each sample was added to the wells and incubated at 37°C for 1 h. About 100 μL of Detection Solution A was added to each well after the liquid was disposed of and the wells were dried. The wells were then incubated for 1 h at 37°C. After discarding the liquid, 100 μL of Detection Solution B was added, and the mixture was incubated for 30 min at 37°C. The optical density (OD value) was measured using a microplate reader (iD3, Molecular Devices, San Jose, CA, USA). A standard curve was plotted, and the concentrations of the samples were calculated.
Human umbilical vein endothelial cell (HUVEC) migration and invasion assay (transwell assay)
We used 24-well plates with transwell chambers with 8 μm pores for cell invasion tests. An initial coating of 50 μL of Matrigel (40182, Yeasen, Shanghai, China) was applied to the transwell’s top chamber. About 200 μL of the cell suspension (2.5×104 cells/mL) was planted into the upper chamber after the transfected NCI–N87 cells had solidified and were resuspended in serum-free basic media. A complete medium with 10% fetal bovine serum was placed into the lower chamber. A cotton swab was used to remove the cells in the upper chamber from the membrane’s upper surface following a 24-h incubation period. Crystal violet solution (C0121, Beyotime Biotechnology, Shanghai, China) was used to stain the cells for 20 min on the chamber’s lower surface after they had been fixed with 4% paraformaldehyde (P0099, Beyotime Biotechnology, Shanghai, China). PBS that had been chilled beforehand was used to wash the cells after 30 min. To determine the invasion capacity based on the cell count, images of the invaded cells were obtained under a microscope, and the quantity of invaded cells was calculated using ImageJ software.
HUVEC tubule formation assay
The concentration of Matrigel was diluted and coated onto a 96-well plate for 1 h. Cells from each group were collected, and 100 μL of the cell suspension was seeded into each well of the 96-well plate. Each group had three replicates, with a total of five plates. For 6 h, the plates were incubated at 37°C with 5% CO2. Under a microscope, the development of HUVEC tubules was observed. For each well, images of three random fields were captured, and the number of tubules was counted. The average number of tubules from the three fields was calculated.
Co-Immunoprecipitation (IP)
Antibodies (14793 or 2622, CST, Ma, BSN, USA) were incubated with protein A/G beads (P2179M, Beyotime Biotechnology, Shanghai, China). For the IP group, 800 μL of ice-cold PBS, 200 μL of RIPA lysis buffer, 30 μL of protein A/G beads, and 2 μg of either RGS4 or MDK antibody were added. For the IgG group, 800 μL of ice-cold PBS, 200 μL of RIPA lysis buffer, 30 μL of protein A/G beads, and 2 μg of the corresponding IgG antibody (3900, CST, Ma, BSN, USA) were added. The mixtures were incubated at 4°C for 8 h with continuous inversion for mixing. Cell protein extraction was performed as described previously for Western blot experiments. The extracted proteins were divided into three portions: One as input and the two other portions added to the incubated beads, with approximately 2 mg of protein in each tube. The mixtures were incubated overnight at 4°C with continuous inversion for mixing. The next day, the beads were washed 5 times with pre-cooled IP wash buffer at low temperatures. About 40 μL of 2.5 × loading buffer (BL529B, Biosharp Life Science, Anhui, China) was added to each tube, mixed thoroughly, and heated on a metal constant temperature mixer for 10 min. After centrifugation at 4°C and 12,000 r/min for 10 min, the supernatant was collected. Western blot experiments were then performed as previously described.
Immunofluorescence staining
Inoculate logarithmic phase cells into a 24-well plate pre-seeded with coverslips (1 × 104 cells/well, 1 mL/well) and incubate at 37°C in a 5% CO2 incubator. Once the cells adhered, they were treated under various conditions as required, and we continued to incubate the 24-well plate at 37°C in a 5% CO2 incubator for 24 h. After removing the 24-well plate and washing it with PBS, we added 1 mL of 4% paraformaldehyde to each well for 30 min of fixation. The plate was washed twice with PBS for 1 min each time. Each well was treated with 1% Triton X-100 (IR9073, Solarbio, Beijing, China) for 15 min, washed twice with PBS for 1 min each time, blocked with 5% skim milk for 30 min, and washed twice with PBS for 1 min each time. We diluted primary antibodies E-cadherin (ab214063) or N-cadherin (ab19348) in PBS at a ratio of 1:200 and incubated them overnight at 4°C. After three washes with PBS for 10 min each time, we diluted Fluorescein Isothiocyanate (FITC)-labeled goat anti-rabbit IgG secondary antibody (ab6717) and Tetramethylrhodamine (TRITC)-labeled goat anti-mouse IgG secondary antibody (ab6786) in PBS at a ratio of 1:100. The sample was incubated at 37°C for 1 h and protected it from light starting from the secondary antibody step. After two washes with PBS for 10 min each time, we added a small amount of glycerol to a slide, removed the coverslip, and mounted it on the slide. Images were captured using a fluorescence microscope. All antibodies were purchased from Abcam (Cambridge, MA, USA).
Real-time quantitative polymerase chain reaction (qPCR) assay
Total ribonucleic acid (RNA) was extracted by TRIzol reagent (R0016, Beyotime Biotechnology, Shanghai, China), and RNA was reverse transcribed into complementary DNA (cDNA) using the Quant cDNA first-strand synthesis kit (KR103, Tiangen, Beijing, China). According to the instructions of the reverse transcription kit, 20 μL of reverse transcription reaction mixture was prepared, and 1 μg of total cDNA was added. Fast-fire SYBR green reagent (FP207, Tiangen, Beijing, China) was used for qPCR to detect the mRNA expression of genes in Table 1 using the above primers. GAPDH gene expression was used as the endogenous control, and the (2−∆∆Ct) ratio was used to calculate the expression level.
| Primer | Primer sequences (5’-3’) |
|---|---|
| RSG4 | F: ACATCGGCTAGGTTTCCTGC |
| R: GTTGTGGGAAGAATTGTGTTCAC | |
| MDK | F: CGCGGTCGCCAAAAAGAAAG |
| R: TACTTGCAGTCGGCTCCAAAC | |
| GAPDH | F: GGAGCGAGATCCCTCCAAAAT |
| R: GGCTGTTGTCATACTTCTCATGG |
F: Forward, R: Reverse, RSG4: Regulator of G protein signaling 4, MDK: Midkine, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
Bioinformatic analysis
The relationship between the expression levels of RGS4 and MDK and the prognosis of patients with GC was analyzed using Kaplan–Meier (KM) survival analysis (http://kmplot. com/analysis/). The TNM plot database (https://tnmplot. com/analysis/) was utilized to analyze the correlation between RGS4 expression and clinicopathological features of lung cancer pathological stages.
Statistical analysis
Statistical analysis was performed using the Statistical Package for the Social Sciences 22.0 software (IBM Corp., Armonk, NY, USA). Normally distributed data were expressed as mean ± standard deviation. Comparisons between groups were performed using independent sample t-tests, and comparisons among multiple groups were conducted using one-way analysis of variance, followed by Tukey’s post hoc test. P < 0.05 was considered statistically significant.
RESULTS
RGS4 is a key factor in GC metastasis
To explore the role of RGS4 in GC metastasis, we first knocked down RGS4 expression in NCI–N87 GC cells through RNA interference technology. Figures 1a and b show that the protein expression level of RGS4 in the RGS4 shRNA (shRGS4) group was significantly reduced (P < 0.001), confirming the effectiveness of RGS4 knockdown. The lung tissues were subjected to H&E staining to observe the formation of lung metastatic lesions. Figure 1c shows that the number of lung metastatic lesions in the shRGS4 group was significantly reduced. Further statistical analysis confirmed the significance of this observation ([Figure 1d], P < 0.001). To further validate the role of RGS4 in GC metastasis, we overexpressed RGS4 in NCI–N87 cells. Figures 1e and f show that the protein level of RGS4 in the RGS4 overexpression group significantly increased (P < 0.001), confirming the effectiveness of overexpression. We similarly injected the RGS4-overexpressing NCI–N87 cells into nude mice through the tail vein. H&E staining of lung tissues after 6 weeks showed that the number of lung metastatic lesions in the RGS4 overexpression group significantly increased [Figure 1g]. Statistical analysis further confirmed the significance of this result, P < 0.01 (Figure 1h). These results collectively indicated that the expression level of RGS4 was positively correlated with the metastatic ability of GC cells. Knockdown of RGS4 significantly inhibited the metastatic ability of GC cells, whereas overexpression of RGS4 significantly enhanced their metastatic potential.
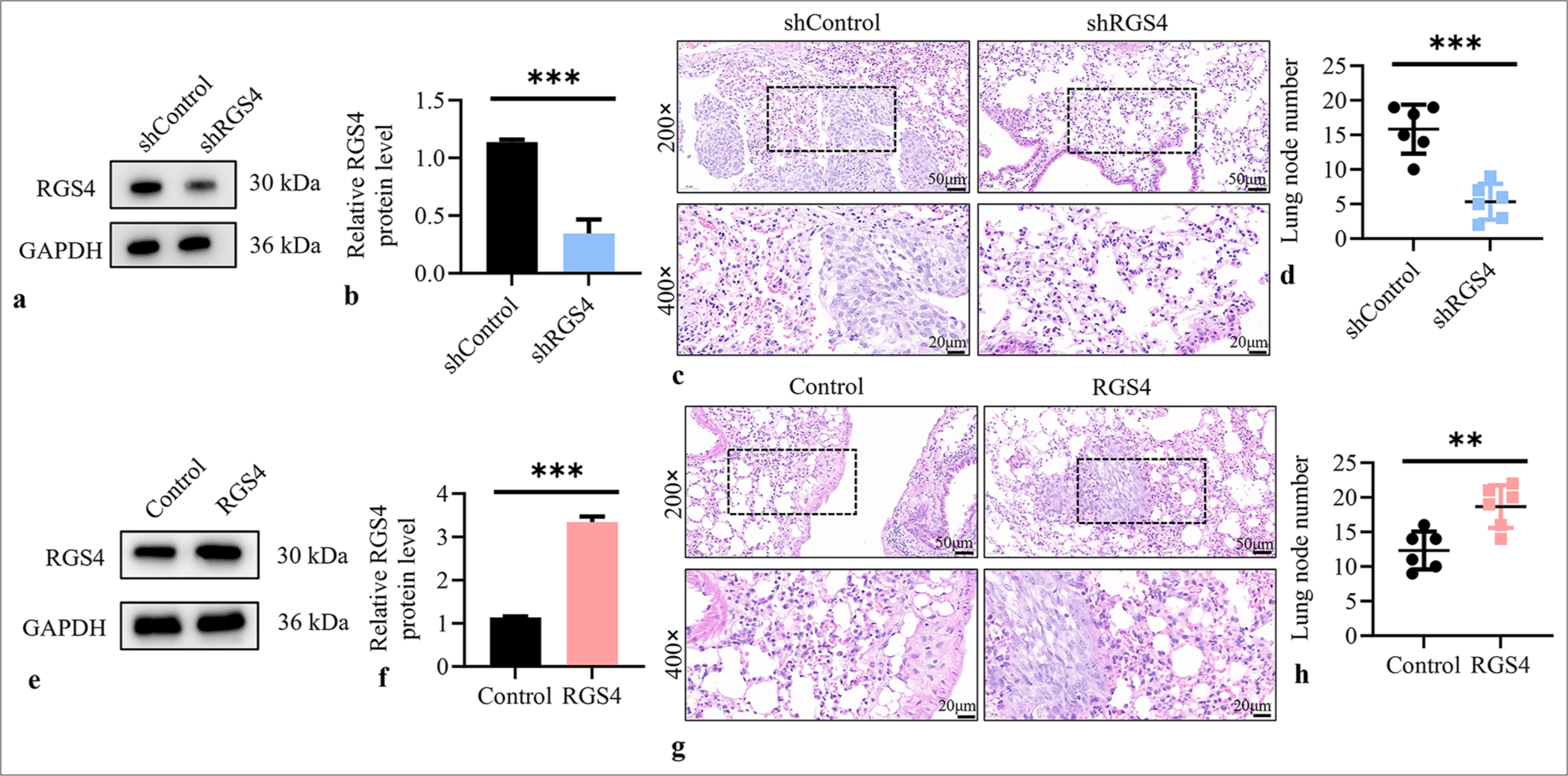
- RGS4 is a key factor in gastric cancer metastasis. (a and b) Validation of transfection efficiency in NCI–N87 cells after transfection with silencing plasmid. (c) Lung tissue was stained with H&E, objective: 200× and the metastases after silencing RGS4 were observed. Scale bar: 50 or 20 μm. (d) Statistical analysis of the number of lung metastatic foci (n = 6). (e and f) Validation of transfection efficiency in NCI– N87 cells after transfection with overexpression plasmid. (g) Lung tissue was stained with H&E, objective: 200× and the metastases after overexpression of RGS4 were observed. Scale bar: 50 or 20 μm. (h) Statistical analysis of the number of lung metastatic foci (n = 6). n = 3. ✶✶P < 0.01, ✶✶✶P < 0.001. RGS4: Regulator of G protein signaling 4, sh-control: Negative control to RGS4 shRNA, shRGS4: RGS4 shRNA, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, RGS4: Regulator of G protein signaling 4, H&E: Hematoxylin and eosin.
RGS4 induces angiogenesis in GC
Figures 2a and b show that overexpression of RGS4 in NCI–N87 GC cells significantly increased the protein expression levels of VEGF-A and VEGF-B but not VEGF-C (P < 0.001). ELISA results indicated that the levels of VEGF-A and VEGF-B in the culture supernatant of the RGS4 overexpression group were significantly higher than those in the control group (P < 0.001), whereas the level of VEGF-C remained unchanged [Figure 2c]. Conversely, when RGS4 was knocked down in NCI–N87 cells, the protein expression levels of VEGF-A and VEGF-B were significantly reduced (P < 0.001), whereas VEGF-C expression remained unchanged [Figure 2d and e]. ELISA further confirmed that the levels of VEGF-A and VEGF-B in the culture supernatant of the RGS4 knockdown group were significantly lower than those in the control group (P < 0.001), with no significant difference in VEGF-C levels [Figure 2f]. To evaluate the effect of RGS4 on endothelial cell function, we treated HUVECs with conditioned media from NCI–N87 cells subjected to RGS4 genetic manipulation. Transwell assays showed that the conditioned media from the RGS4 overexpression group significantly promoted the migration (P < 0.01) and invasion (P < 0.05) abilities of HUVECs [Figure 2g and h]. By contrast, conditioned media from the RGS4 knockdown group significantly inhibited the migration and invasion of HUVECs (P < 0.001) [Figure 2i and j]. In addition, we conducted tube formation assays to assess the effect of RGS4 on angiogenesis. The results showed that the conditioned media from the RGS4 overexpression group significantly enhanced the tube formation ability of HUVECs (P < 0.05) [Figure 2k and l]. By contrast, conditioned media from the RGS4 knockdown group markedly inhibited the tube formation of HUVECs (P < 0.001) [Figure 2m and n]. These experimental results clearly demonstrated that RGS4 could upregulate the expression and secretion of VEGF-A and VEGF-B in GC cells, thereby promoting the migration, invasion, and tube formation abilities of endothelial cells.
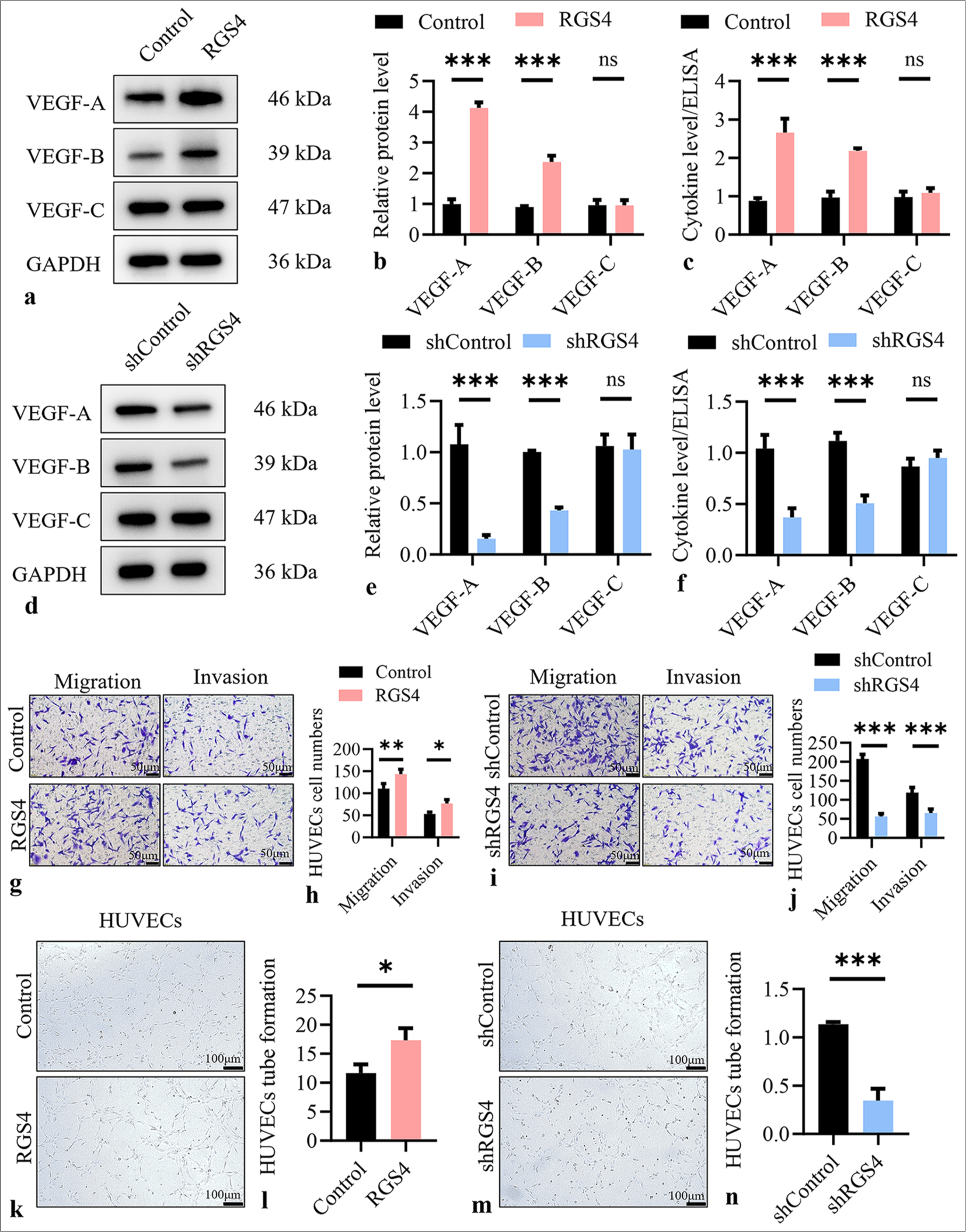
- RGS4 induces angiogenesis in gastric cancer. (a and b) Relative expression of VEGF-A, VEGF-B, and VEGF-C in NCI–N87 cells overexpressing RGS4. (c) Relative level of VEGF-A, VEGF-B, and VEGF-C in the supernatant of NCI–N87 cells overexpressing RGS4. (d and e) Relative expression of VEGF-A, VEGF-B, and VEGF-C in RGS4 knockdown NCI–N87 cells. (f) Relative level of VEGF-A, VEGF-B, and VEGF-C in the supernatant of RGS4 knockdown NCI–N87 cells. (g and h) Migration and invasion assays, objective: 200×, of HUVECs treated with the supernatant of NCI–N87 cells overexpressing RGS4. Scale bar: 50 μm. (i and j) Migration and invasion assays, objective: 200×, of HUVECs treated with the supernatant of RGS4 knockdown NCI–N87 cells. Scale bar: 50 μm. (k and l) Tube formation assay of HUVECs treated with the supernatant of NCI–N87 cells overexpressing RGS4. Scale bar: 100 μm. (m and n) Tube formation assay of HUVECs treated with the supernatant of RGS4 knockdown NCI–N87 cells. Scale bar: 100 μm. n = 3. ns: No significant, ✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001. VEGF: Vascular endothelial growth factor, HUVECs: Human umbilical vein endothelial cells, RGS4: Regulator of G protein signaling 4.
RGS4 interacts with MDK
To investigate the potential interaction between RGS4 and MDK, we conducted a series of protein interaction experiments. First, we co-expressed RGS4-Flag and MDKGlutathione-S-transferase (GST) fusion proteins in cells. Using immunoprecipitation, we precipitated RGS4-Flag with an anti-Flag antibody, resulting in the co-precipitation of MDKGST [Figure 3a]. This result indicated that RGS4 could form a complex with MDK. To further validate this interaction, we performed a GST pull-down assay. We precipitated MDK-GST using glutathione-sepharose 4B beads, and RGS4-Flag was specifically pulled down by MDK-GST [Figure 3b]. This result further confirmed the interaction between RGS4 and MDK. Notably, in both experiments, we did not observe non-specific binding when RGS4-Flag and MDK-GST were expressed individually, which further supported the specificity of the interaction between RGS4 and MDK.
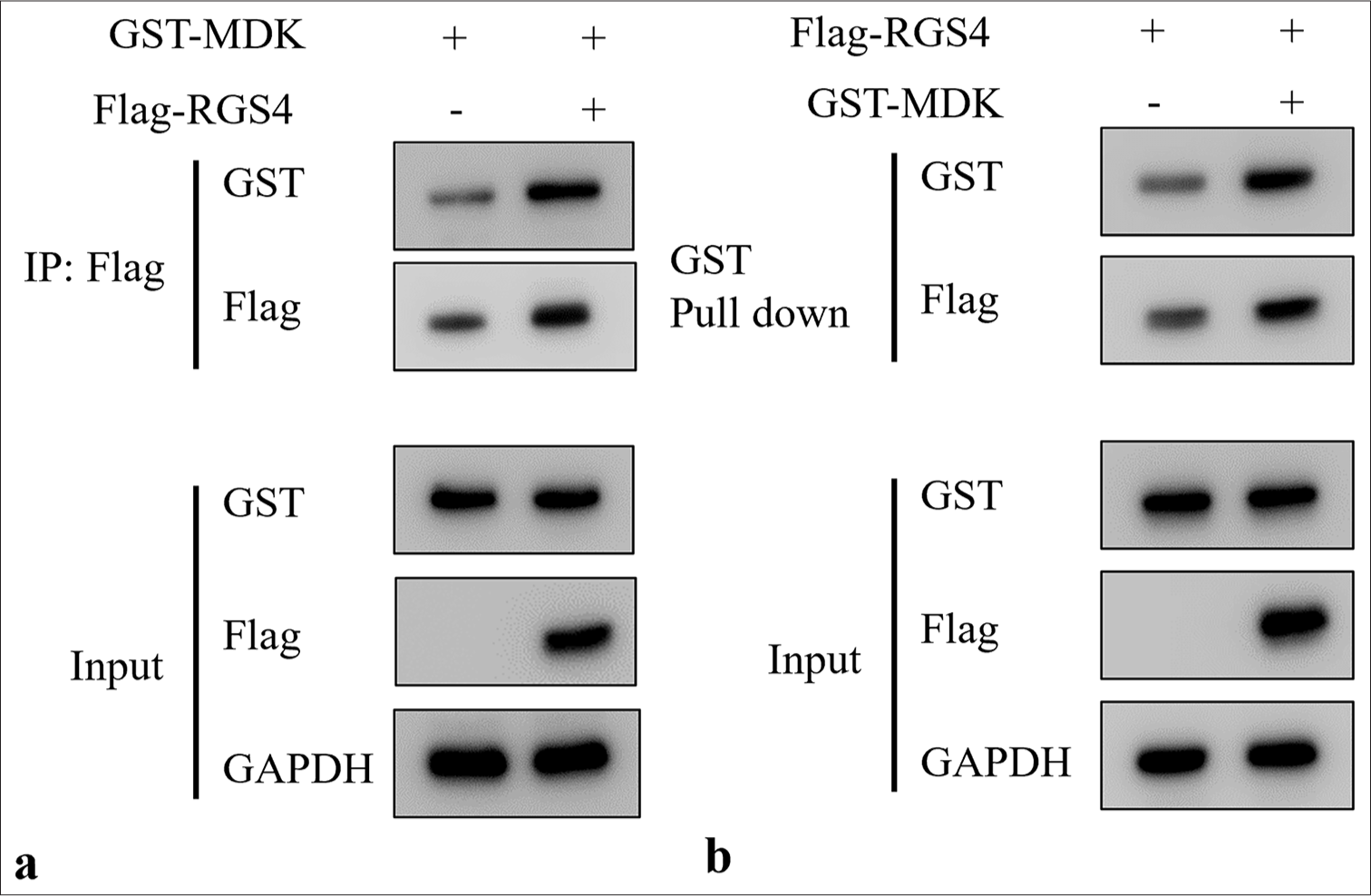
- Interaction between RGS4 and MDK. (a) Co-transfection of HEK293T cells with RGS4-Flag and MDK-GST plasmids, followed by immunoprecipitation of RGS4-Flag using anti-Flag antibody. (b) Co-transfection of HEK293T cells with RGS4-Flag and MDK-GST plasmids, followed by precipitation of MDK-GST using glutathione-Sepharose 4B beads. n = 3. GST: Glutathione-Stransferase, MDK: Midkine, RGS4: Regulator of G protein signaling 4.
RGS4 upregulates the expression of MDK
To explore the regulatory effect of RGS4 on MDK expression, we performed a series of gene manipulation and expression analysis experiments in NCI–N87 GC cells. First, we stably overexpressed RGS4 in NCI–N87 cells. Figure 4a-c shows that the protein and mRNA levels of RGS4 significantly increased in the RGS4 overexpression group (P < 0.01), and the protein levels of MDK were also notably elevated (P < 0.05). This initial result suggested that RGS4 may positively regulate MDK expression. To further validate this observation, we knocked down RGS4 expression in NCI– N87 cells. Figure 4d-f revealed that the protein and mRNA levels of RGS4 were significantly reduced in the shRGS4 group (P < 0.001); correspondingly, the protein levels of MDK considerably decreased (P < 0.001). These findings further supported the positive regulatory role of RGS4 on MDK expression. To visually observe the effect of RGS4 on MDK expression at the single-cell level, we conducted immunofluorescence experiments. In RGS4-overexpressing NCI–N87 cells, we observed a significant increase in the fluorescence intensity of MDK (P < 0.001), indicating elevated expression levels of MDK [Figure 4g and h]. Conversely, in RGS4-knockdown NCI–N87 cells, the fluorescence intensity of MDK was markedly reduced (P < 0.001) [Figure 4i and j], further confirming that RGS4 knockdown led to decreased MDK expression. These experimental results consistently demonstrated that RGS4 could positively regulate MDK expression. Overexpression of RGS4 led to an increase in MDK protein levels, whereas knockdown of RGS4 resulted in a reduction in MDK protein levels.
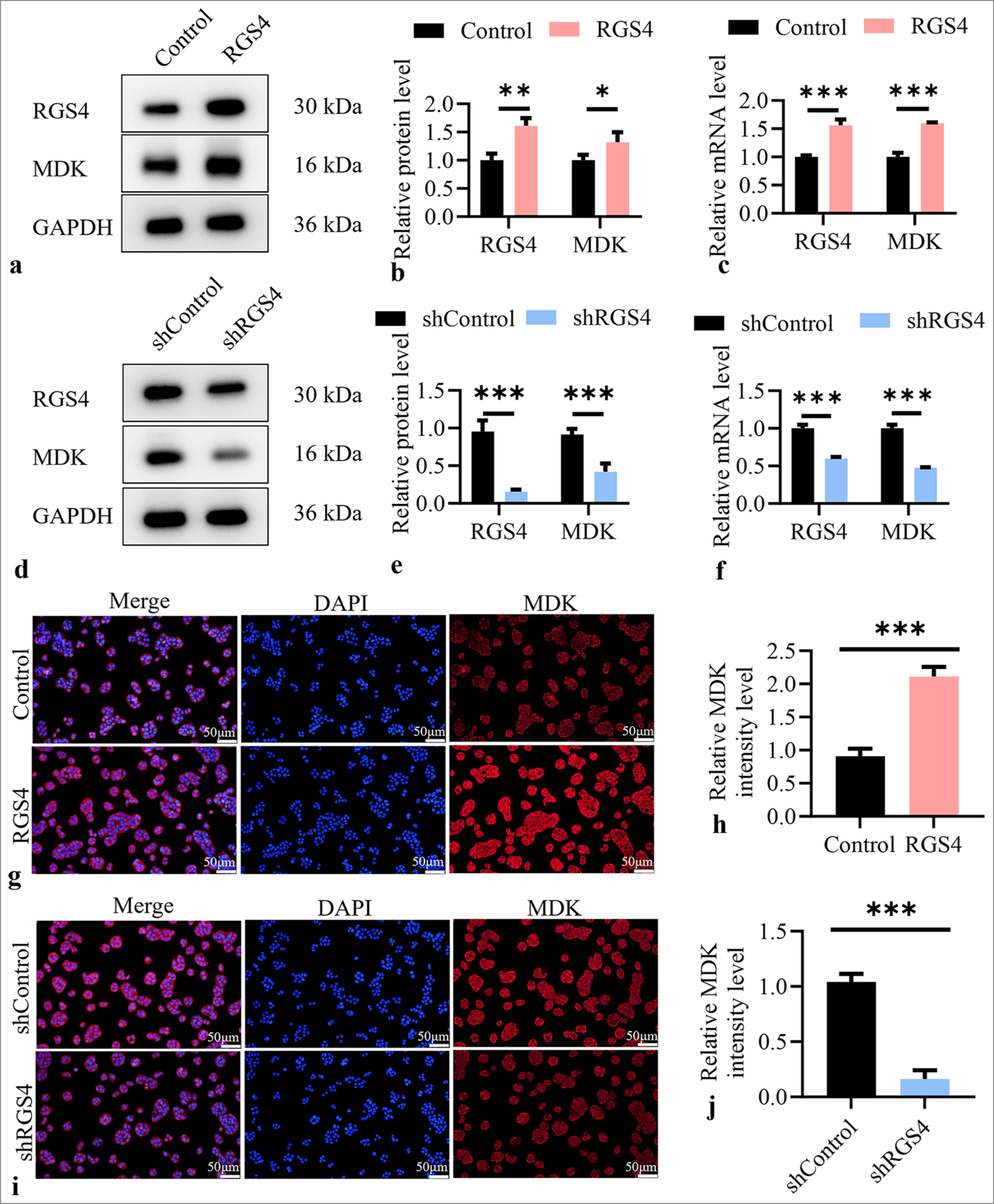
- RGS4 upregulates MDK expression. (a and b) Stable overexpression of RGS4 in NCI–N87 cells, with immunoblot showing protein levels of MDK and RGS4. (c) The mRNA levels of RGS4 and MDK in NCI–N87 cells stabilized with overexpression of RGS4. (d and e) Knockdown of RGS4 in NCI–N87 cells, with immunoblot showing protein levels of MDK and RGS4. Scale bar: 50 μm. (f) The mRNA levels of RGS4 and MDK in NCI–N87 cells that silence RGS4. (g and h) Stable overexpression of RGS4 in NCI–N87 cells, with immunofluorescence, objective: 200×, showing MDK expression levels. Scale bar: 50 μm. (i and j) Knockdown of RGS4 in NCI–N87 cells, with immunofluorescence, objective: 200×, showing MDK expression levels. n = 3. ✶✶✶P < 0.001. DAPI: 4’,6-Diamidino-2’-phenylindole, RGS4: Regulator of G protein signaling 4, MDK: Midkine.
RGS4 inhibits EMT in GC
Immunofluorescence double staining was conducted to investigate the association between RGS4 and EMT in GC. Figures 5a and b show that the overexpression of RGS4 significantly upregulated the fluorescence intensity signal of N-cadherin and significantly downregulated the fluorescence intensity signal of E-cadherin (P < 0.001). In Figures 5c and d, after silencing RSG4, the fluorescence intensity of E-cadherin was significantly upregulated, whereas that of N-cadherin significantly decreased (P < 0.01).
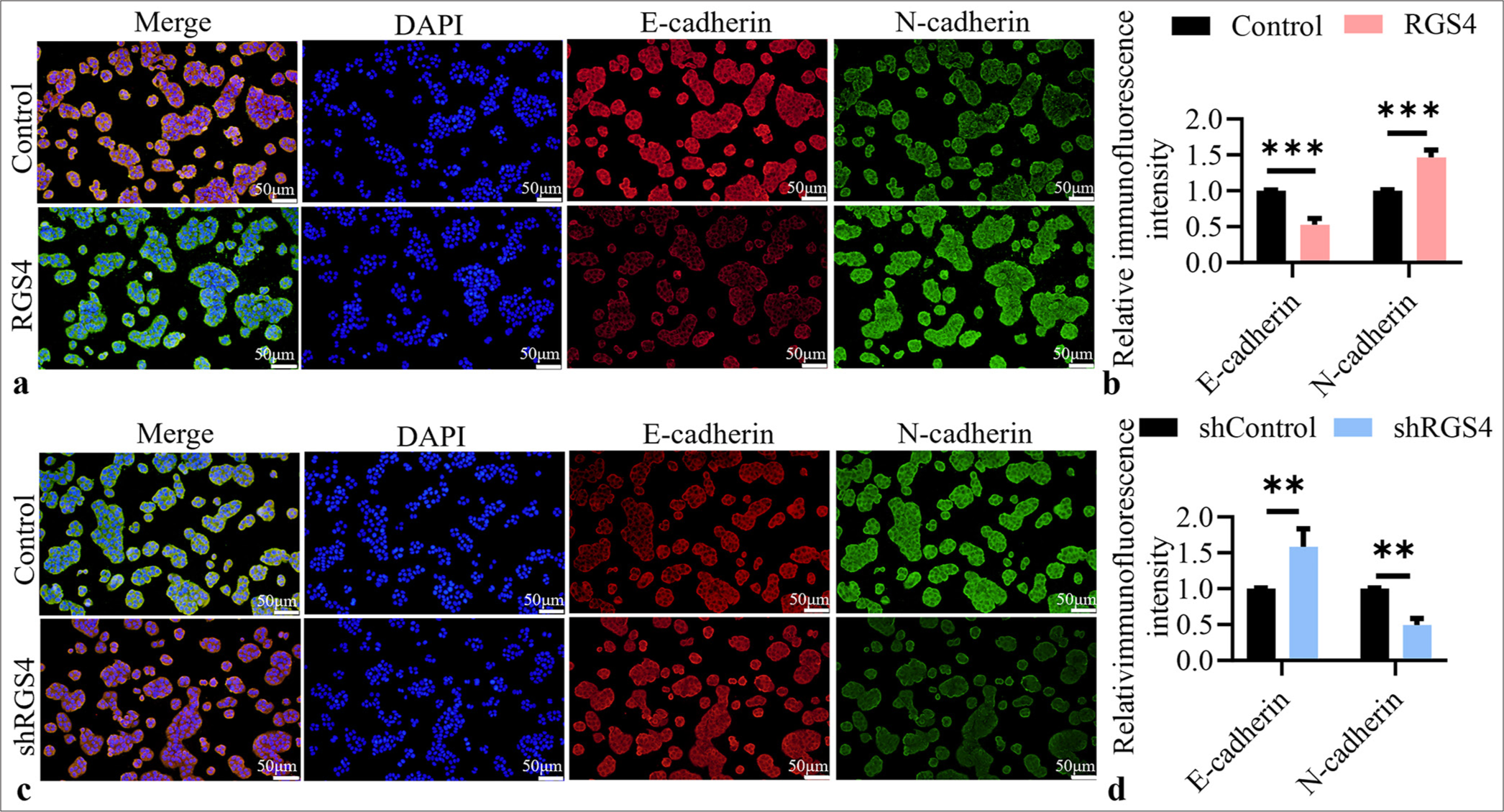
- RGS4 inhibits EMT in GC. (a and b) Immunofluorescence double staining, objective: 200×, of E-cadherin and N-cadherin in NCI– N87 cells after RSG4 overexpression. Scale bar: 50 μm. (c and d) Immunofluorescence double staining, objective: 200×, of E-cadherin and N-cadherin in NCI–N87 cells after silencing RSG4. Scale bar: 50 μm. n = 3. ✶✶P < 0.01, ✶✶✶P < 0.001. RGS4: Regulator of G protein signaling 4, GC: Gastric cancer.
RGS4 promotes angiogenesis in GC by upregulating MDK
To verify whether RGS4 promotes angiogenesis in GC by upregulating MDK, we designed a series of rescue and reverse verification experiments. First, we overexpressed MDK in RGS4 knockdown NCI–N87 cells. Western blot results showed that RGS4 and MDK protein levels were significantly reduced in the shRGS4 group (P < 0.01). However, in the shRGS4+MDK group, MDK expression significantly increased (P < 0.01, Figure 6a and b). This result indicated that MDK overexpression effectively rescued the MDK downregulation caused by RGS4 knockdown. Subsequently, we evaluated the effects of these cell culture supernatants on the function of HUVECs. Transwell assay results showed that the shRGS4 group significantly inhibited the migration and invasion abilities of HUVECs compared with the sh-control group (P < 0.01). However, the shRGS4+MDK group restored HUVEC migration and invasion to normal levels (P < 0.01) [Figure 6c and d]. Similarly, tube formation assays showed that the shRGS4 group significantly reduced the tube formation ability of HUVECs (P < 0.01), whereas the shRGS4+MDK group restored this ability [Figure 6e and f]. These results suggested that MDK overexpression could rescue the angiogenesis inhibition effect caused by RGS4 knockdown. To further validate the mechanism by which RGS4 promotes angiogenesis through MDK, we knocked down MDK in RGS4 overexpressing NCI–N87 cells. Figures 6g and h show that the RGS4 and MDK protein levels significantly increased in the RGS4 group (P < 0.01). However, in the RGS4 + MDK shRNA (shMDK) group, MDK expression was significantly reduced despite RGS4 remaining at high levels. Subsequent functional experiments further supported this mechanism. Transwell assays showed that the RGS4 group significantly promoted the migration and invasion abilities of HUVECs (P < 0.01). However, this promoting effect was completely suppressed in the RGS4 + shMDK group, reducing HUVEC migration and invasion to levels comparable to the control group [Figure 6i and j]. Tube formation assays showed a similar trend: The RGS4 group significantly enhanced the tube formation ability of HUVECs (P < 0.001), whereas the RGS4 + shMDK group completely inhibited this effect [Figure 6k and l].
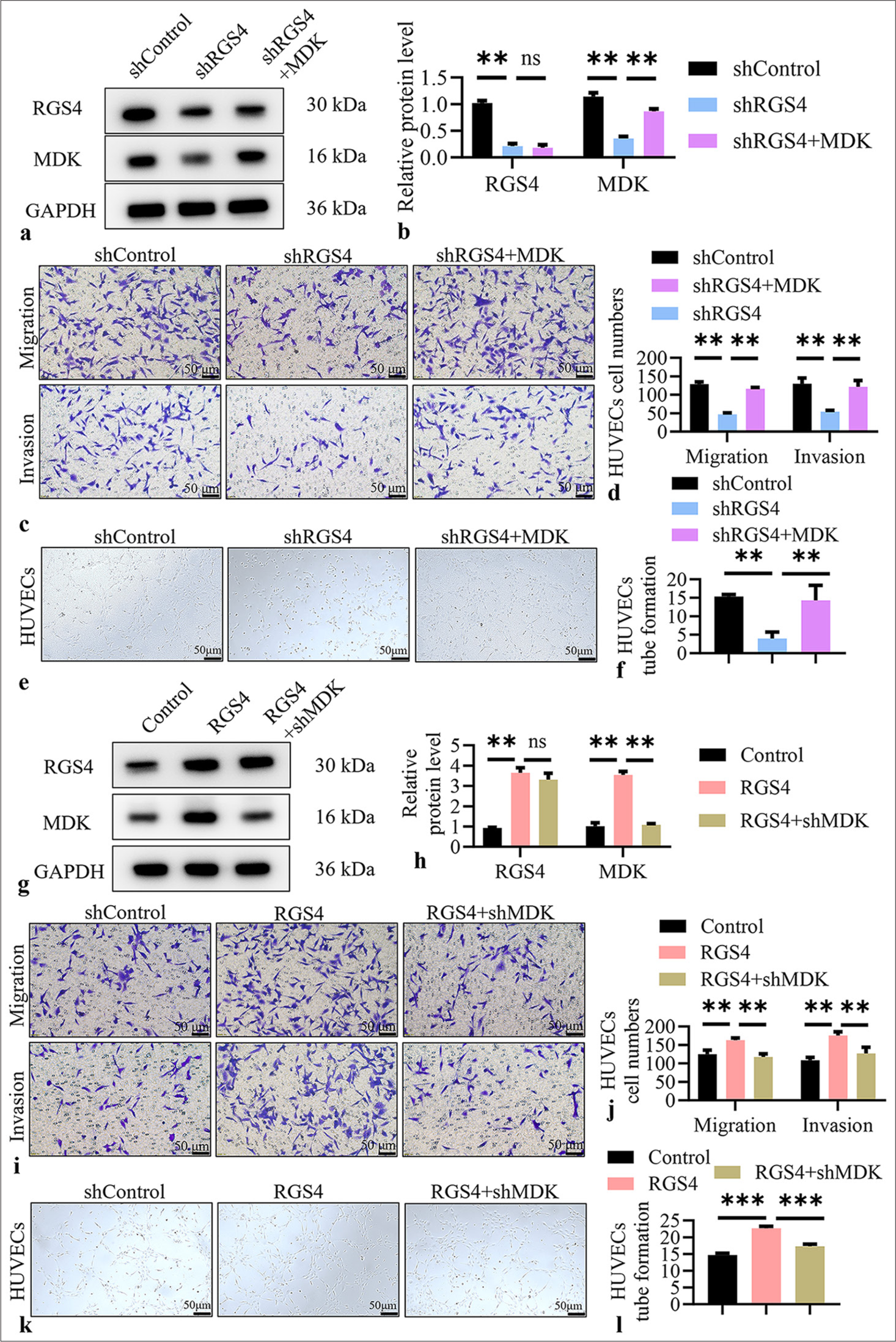
- RGS4 promotes angiogenesis in gastric cancer by upregulating MDK. (a and b) In NCI–N87 cells with RGS4 knockdown, transfection with MDK and control vectors, with immunoblot showing protein levels of MDK and RGS4. (c and d) Migration assay, objective: 200×, to analyze HUVEC migration and invasion capabilities. Scale bar: 50 μm. (e and f) Tube formation assay, objective: 200×, to analyze HUVEC angiogenesis. Scale bar: 50 μm. (g and h) Knockdown of MDK in NCI–N87 cells overexpressing RGS4, with immunoblot showing protein levels of MDK and RGS4. (i and j) Migration assay, objective: 200×, to analyze HUVEC migration and invasion capabilities. Scale bar: 50 μm. (k and l) Tube formation assay, objective: 200×, to analyze HUVEC angiogenesis. Scale bar: 50 μm. n = 3. ns: No significant, ✶✶P < 0.01, ✶✶✶P < 0.001. shMDK: MDK shRNA, RGS4: Regulator of G protein signaling 4, MDK: Midkine, HUVECs: Human umbilical vein endothelial cells.
RGS4 promotes GC metastasis by upregulating MDK
To further validate the role of RGS4 in promoting GC metastasis by upregulating MDK, we conducted in vivo experiments. We injected RGS4 knockdown NCI–N87 cells (shRGS4), RGS4 knockdown cells with MDK overexpression (shRGS4 + MDK), and control cells (shControl) into the tail veins of BALB/c nude mice to establish a lung metastasis model. After 6 weeks, we collected and analyzed the lung tissues of the nude mice. First, we observed lung metastatic nodules by gross examination and H&E staining [Figure 7a]. The results showed that the number of lung metastatic nodules was significantly reduced in the shRGS4 group (P < 0.01). However, the number of lung metastatic nodules in the shRGS4 + MDK group was restored to normal levels (P < 0.01). This result indicated that MDK overexpression could rescue the metastasis inhibition effect caused by RGS4 knockdown. To precisely quantify this phenomenon, we performed statistical analysis on the lung metastatic nodules [Figure 7b]. The results further confirmed our observation: The number of lung metastatic nodules in the shRGS4 group was significantly lower, whereas the number of lung metastatic nodules in the shRGS4+MDK group did not differ significantly from that in the sh-control group (P < 0.01). This quantitative analysis reinforced the critical role of MDK in the RGS4-mediated promotion of GC metastasis. Given the importance of angiogenesis in tumor metastasis, we also performed CD31 immunohistochemical staining on the lung metastatic nodules [Figure 7c and d]. CD31, a marker of endothelial cells, reflects the extent of tumor angiogenesis. The results showed that the number of CD31-positive cells was significantly reduced in the shRGS4 group (P < 0.001), indicating inhibited angiogenesis. However, the number of CD31-positive cells in the shRGS4+MDK group was restored to levels comparable to the sh-control group (P < 0.001). This result indicated that MDK overexpression could rescue the angiogenesis inhibition effect caused by RGS4 knockdown, thereby promoting tumor metastasis. In summary, these in vivo experiments provided strong evidence that RGS4 promotes GC metastasis by upregulating MDK. We observed that RGS4 knockdown significantly inhibited the pulmonary metastatic capability of GC cells, whereas MDK overexpression effectively reversed this inhibition. Notably, we found that this process was closely related to tumor angiogenesis, further supporting the critical role of the RGS4– MDK axis in promoting GC angiogenesis and metastasis.
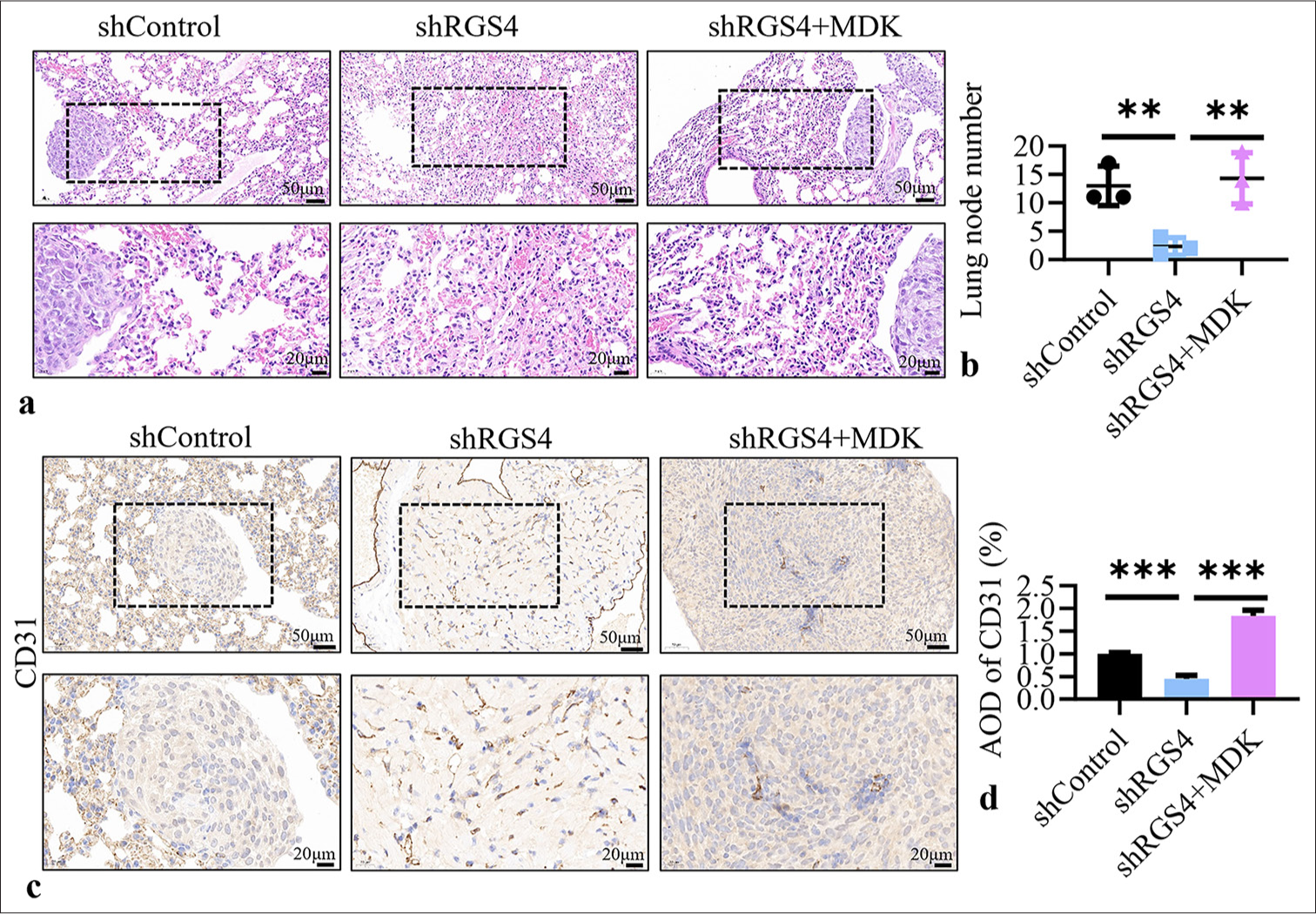
- RGS4 promotes gastric cancer tumor metastasis by upregulating MDK. (a) In NCI–N87 cells with RGS4 knockdown, transfection with MDK and control vectors, followed by detection of lung metastatic nodules in mice 6 weeks after tail vein injection. Scale bar: 50 or 20 μm. (b) Statistical analysis of the number of lung metastatic foci. (c and d) Immunohistochemical detection of CD31 in lung metastatic nodules. Scale bar: 50 or 20 μm. n = 3. ✶✶P < 0.01, ✶✶✶P < 0.001. RGS4: Regulator of G protein signaling 4, MDK: Midkine.
High expression of RGS4 and MDK predicts poor prognosis in GC patients
To evaluate the relationship between the expression levels of RGS4 and MDK and the clinical prognosis of GC patients, we conducted survival analysis using the KM plotter database. This database included extensive gene expression data and clinical follow-up information for GC patients, enabling us to perform reliable prognostic analysis. We focused on three key survival metrics: First progression (FP), overall survival (OS), and post-progression survival (PPS). First, we analyzed the relationship between RGS4 expression levels and patient prognosis [Figure 8a-c]. The results showed that GC patients with high RGS4 expression had significantly poorer prognoses across all three survival metrics compared with their counterparts. Second, we performed a similar analysis for MDK expression levels and patient prognosis [Figure 8d-f]. The results also indicated that GC patients with high MDK expression exhibited poorer prognoses across all three survival metrics compared with their counterparts. Our survival analysis results strongly confirmed the close association between high expression of RGS4 and MDK and poor prognosis in GC patients. Figures 8g and h show that the expression level of RGS4 in GC was positively correlated with the tumor T stage. These findings highlight the potential value of RGS4 and MDK as prognostic biomarkers and provide clinical rationale for targeting the RGS4–MDK axis in GC therapy.
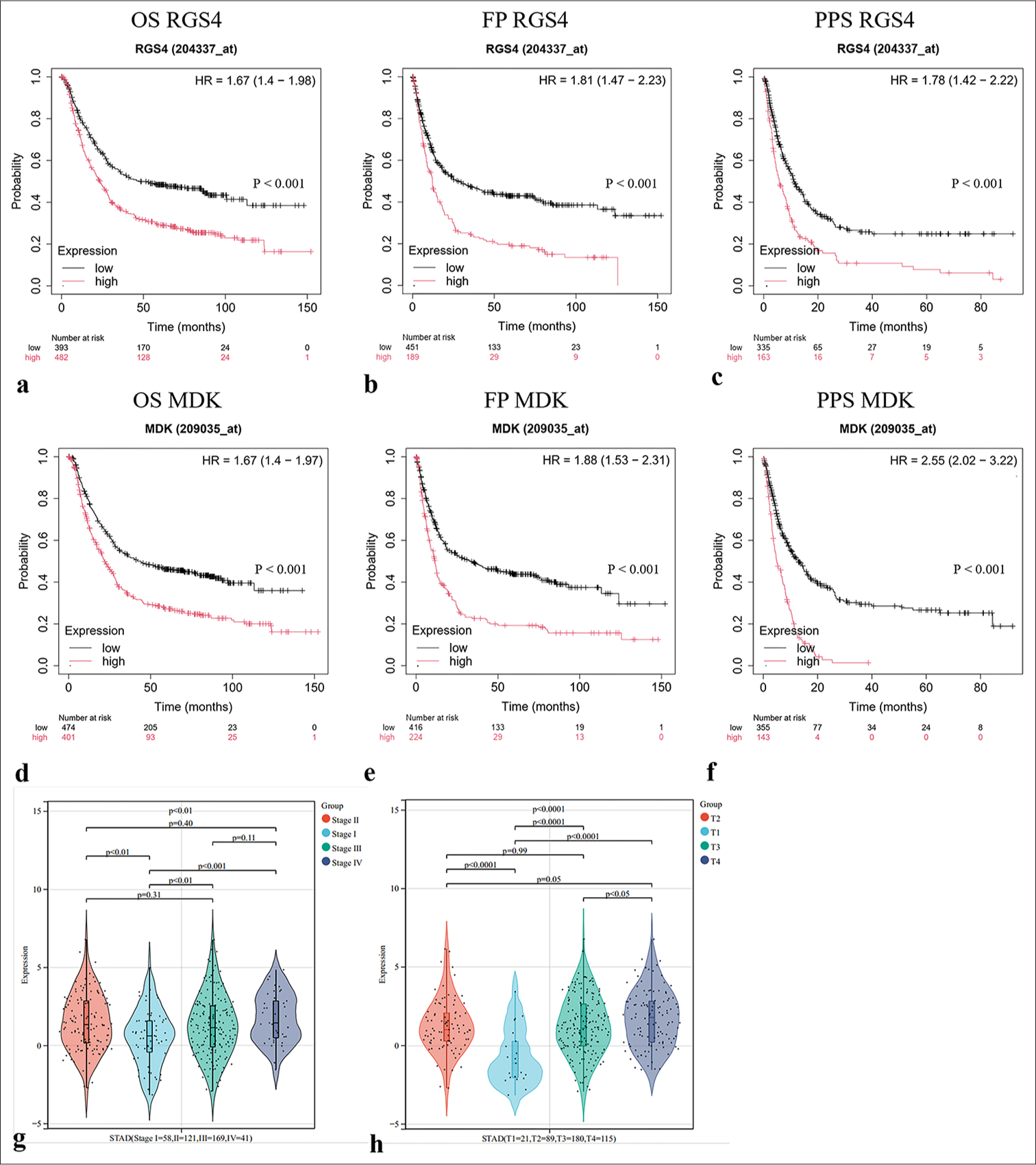
- High expression of RGS4 and MDK predicts poor prognosis in gastric cancer patients. (a-f) Analysis of the correlation between RGS4 or MDK expression and patient outcomes (FP: First progression, OS: Overall survival, PPS: Post-progression survival) in gastric cancer using the KM database (http://kmplot.com/analysis/). (g and h) Correlation analysis of RSG4 expression levels with lung cancer stages. FP: First progression, OS: Overall survival, PPS: Post-progression survival, STAD: Stomach adenocarcinoma, RGS4: Regulator of G protein signaling 4, MDK: Midkine, KM: Kaplan–Meier.
DISCUSSION
In this investigation, we discovered a substantial correlation between the invasiveness and angiogenic potential of tumors and the expression level of RGS4. Analysis of GC samples revealed that high expression of RGS4 was closely associated with poor prognosis. This finding was consistent with previous studies on other types of cancers. For instance, RGS4 is intimately linked to tumor invasion and metastasis and is significantly expressed in prostate and breast cancer.[13,15,21] These results suggested that RGS4 may play a similar role in promoting tumor progression across multiple cancers.
Through in vitro and in vivo experiments, we further confirmed the critical role of RGS4 in GC angiogenesis. In vitro experiments demonstrated that overexpression of RGS4 significantly enhanced the angiogenic capacity of GC cells, whereas knockdown of RGS4 inhibited this process. In vivo studies supported these findings, as nude mouse models with RGS4 overexpression exhibited more lung metastases compared with those with RGS4 knockdown, which showed fewer lung metastases. These results indicated that RGS4 played a key role in promoting GC angiogenesis and metastasis. Further mechanistic studies revealed that RGS4 promoted angiogenesis in GC by upregulating MDK expression. MDK is a known pro-angiogenic factor with various biological functions, including promoting cell proliferation, migration, and angiogenesis.[22] In our study, RGS4 overexpression significantly upregulated MDK expression, whereas knockdown of RGS4 downregulated MDK expression. Immunoprecipitation and immunofluorescence co-staining experiments showed that RGS4 and MDK were co-localized within cells, and RGS4 could directly interact with MDK. These results suggested that RGS4 may promote GC angiogenesis by directly regulating the expression and function of MDK. To further verify the role of RGS4 in MDK-mediated angiogenesis, we conducted supplementary experiments. Overexpression of MDK in RGS4 knockdown GC cells partially restored their angiogenic capability, whereas knockdown of MDK in RGS4-overexpressing GC cells significantly inhibited their angiogenic capacity. These results further support the hypothesis that RGS4 promotes GC angiogenesis by upregulating MDK expression.
Our findings have important clinical implications. First, the high expression levels of RGS4 and MDK were closely associated with poor prognosis in GC patients, suggesting that they could serve as potential prognostic markers. Second, the critical roles of RGS4 and MDK in GC angiogenesis indicated that they may be potential therapeutic targets. Targeted inhibition of RGS4 or MDK expression or function may help suppress angiogenesis and metastasis in GC, thereby improving patient prognosis.
However, this study had several limitations.[23,24] First, our research primarily focused on the role of RGS4 and MDK in GC angiogenesis without a comprehensive investigation of their roles in other tumor-related biological behaviors. Second, our study was mainly based on in vitro and in vivo experimental models, and it is unclear whether the results of this study are applicable to clinical samples. Furthermore, the specific regulatory mechanisms of RGS4 and MDK require further in-depth research to elucidate their overall roles in GC. Future research can further explore the roles and mechanisms of RGS4 and MDK in GC from the following aspects. First, large-scale clinical sample analyses can be conducted to verify the expression levels of RGS4 and MDK in GC patients and their relationship with prognosis. Second, gene editing techniques can be used to construct knockout or overexpression models of RGS4 and MDK in nude mice to investigate their specific roles in the initiation and progression of GC. Finally, high-throughput screening can be employed to identify specific inhibitors targeting RGS4 and MDK and evaluate their potential in GC therapy.
SUMMARY
This study is the first to reveal the critical role of RGS4 in GC angiogenesis and elucidate its molecular mechanism through the upregulation of MDK expression. Our findings not only enrich our understanding of the mechanisms of GC angiogenesis but also provide potential targets for developing new anti-angiogenic therapies. By gaining a thorough understanding of the functions and mechanisms of RGS4 and MDK in GC, we hope to offer effective treatment strategies for patients to improve their prognosis. Future research will further uncover the comprehensive roles of RGS4 and MDK in GC and explore their potential applications in clinical therapy.
AVAILABILITY OF DATA AND MATERIALS
The datasets and materials used and/or analyzed during the current study were available from the corresponding author on reasonable request.
ABBREVIATIONS
ECL: Enhanced chemiluminescence
FP: First Progression
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
GC: Gastric cancer
H&E: Hematoxylin and eosin
MDK: Midkine
OS: Overall Survival
PBS: Phosphate buffer saline
PPS: Post-Progression Survival
RGS4: Regulator of G protein signaling 4
STAD: Stomach adenocarcinoma
ACKNOWLEDGMENT
Not applicable.
AUTHOR CONTRIBUTIONS
YXH and KL: Designed the study; all authors conducted the study; HPS and HL: Collected and analyzed the data; HPS and YXH: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, took public responsibility for appropriate portions of the content, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work were appropriately investigated and resolved.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Qingdao Central Hospital, University of Health and Rehabilitation Sciences (approval number [Y]KY201805802), 2018.10.15. The study was approved by the Institutional Animal Care and Use Committee of Qingdao Central Hospital, University of Health and Rehabilitation Sciences. Because this study only involved animal experiments, informed consent was not applicable
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: Not applicable.
References
- Surgical management of gastric cancer: A review. JAMA Surg. 2022;157:446-54.
- [CrossRef] [PubMed] [Google Scholar]
- Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264-79.
- [CrossRef] [PubMed] [Google Scholar]
- Global burden of gastric cancer: Epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. 2023;20:338-49.
- [CrossRef] [PubMed] [Google Scholar]
- Current developments in gastric cancer: From molecular profiling to treatment strategy. Nat Rev Gastroenterol Hepatol. 2023;20:155-70.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of gastric cancer stem cell on gastric cancer invasion, migration and angiogenesis. Int J Med Sci. 2020;17:2040.
- [CrossRef] [PubMed] [Google Scholar]
- Angiogenesis is associated with an attenuated tumor microenvironment, aggressive biology, and worse survival in gastric cancer patients. Am J Cancer Res. 2021;11:1659.
- [Google Scholar]
- Treatment strategies of gastric cancer-molecular targets for anti-angiogenic therapy: A state-of-theart review. J Gastrointest Cancer. 2021;52:476-88.
- [CrossRef] [PubMed] [Google Scholar]
- The regulatory mechanism and biological significance of the Snail-miR590-VEGFR-NRP1 axis in the angiogenesis, growth and metastasis of gastric cancer. Cell Death Dis. 2020;11:241.
- [CrossRef] [PubMed] [Google Scholar]
- Gastric cancer: A comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 2020;39:1179-203.
- [CrossRef] [PubMed] [Google Scholar]
- JP3, an antiangiogenic peptide, inhibits growth and metastasis of gastric cancer through TRIM25/SP1/MMP2 axis. J Exp Clin Cancer Res. 2020;39:118.
- [CrossRef] [PubMed] [Google Scholar]
- Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 2009;69:5743-51.
- [CrossRef] [PubMed] [Google Scholar]
- RGS4 regulates proliferation and apoptosis of NSCLC cells via microRNA-16 and brain-derived neurotrophic factor. Onco Targets Ther. 2019;12:8701-14.
- [CrossRef] [PubMed] [Google Scholar]
- RGS6 suppresses TGF-β-induced epithelial-mesenchymal transition in non-small cell lung cancers via a novel mechanism dependent on its interaction with SMAD4. Cell Death Dis. 2022;13:656.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting RGS4 ablates glioblastoma proliferation. Int J Mol Sci. 2020;21:3300.
- [CrossRef] [PubMed] [Google Scholar]
- Midkine (MDK) growth factor: A key player in cancer progression and a promising therapeutic target. Oncogene. 2020;39:2040-54.
- [CrossRef] [PubMed] [Google Scholar]
- Regulation of MDK expression in human cancer cells modulates sensitivities to various anticancer drugs: MDK overexpression confers to a multi-drug resistance. Cancer Lett. 2007;247:40-7.
- [CrossRef] [PubMed] [Google Scholar]
- IDDF2022-ABS-0039 Icaritin inhibits tumor metastasis by suppressing MDK-notch signaling pathway in hepatocellular carcinoma. Gut. 2022;71(Suppl 2):A14.
- [CrossRef] [Google Scholar]
- Midkine (MDK) in hepatocellular carcinoma: More than a biomarker. Cells. 2024;13:136.
- [CrossRef] [PubMed] [Google Scholar]
- Exosomal miR-9 inhibits angiogenesis by targeting MDK and regulating PDK/AKT pathway in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2018;37:147.
- [CrossRef] [PubMed] [Google Scholar]
- Function and regulation of RGS family members in solid tumours: A comprehensive review. Cell Commun Signal. 2023;21:316.
- [CrossRef] [PubMed] [Google Scholar]
- MDK induces temozolomide resistance in glioblastoma by promoting cancer stem-like properties. Am J Cancer Res. 2022;12:4825.
- [Google Scholar]
- Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Sig Transduct Target Ther. 2023;8:198.
- [CrossRef] [PubMed] [Google Scholar]
- Reassessing vascular endothelial growth factor (VEGF) in anti-angiogenic cancer therapy. Cancer Treat Res Commun. 2022;32:100620.
- [CrossRef] [PubMed] [Google Scholar]




