


Translate this page into:
Immunoglobulin superfamily member 1 upregulates myc proto-oncogene to accelerate invasion and metastasis of endometrial cancer: Molecular mechanisms and therapeutic prospects

*Corresponding author: Shuai Dong, Department of Gynaecology and Obstetrics, The 960th Hospital of the Joint Logistics Support Force of the People`s Liberation Army of China, Jinan, China. ziguoguoguuwm@163.com
-
Received: ,
Accepted: ,
How to cite this article: Wei J, Jiang J, Zhang S, Dong S. Immunoglobulin superfamily member 1 upregulates myc proto-oncogene to accelerate invasion and metastasis of endometrial cancer: Molecular mechanisms and therapeutic prospects. CytoJournal. 2024; 21:49. doi: 10.25259/Cytojournal_81_2024
Abstract
Objective:
Endometrial cancer (EC) is a common gynecological malignancy, and its metastasis is one of the primary causes of treatment failure. Immunoglobulin superfamily member 1 (IGSF1), a membrane protein, has been associated with the aggressiveness and metastatic capability of various cancers. However, the role and mechanism of this protein in EC remains unclear. Therefore, this study aimed to explore the role of IGSF1 in EC and its possible mechanism.
Material and Methods:
In this study, IGSF1 expression was knocked down through small interfering RNA and short hairpin RNA techniques, and its levels were controlled through overexpression experiments to observe its effects on Ishikawa cells. Wound healing assays, Transwell migration and invasion assays, quantitative real-time polymerase chain reaction, Western blot, and immunofluorescence double labeling were performed to evaluate the ability of cells to migrate, invade, and express markers of the epithelium mesenchymal transition (EMT). In addition, we investigated the regulatory role of IGSF1 in Myc proto-oncogene (c-Myc) expression and its function in lung metastasis through animal models of lung metastasis.
Results:
The results indicate that IGSF1 knockdown inhibited EMT and greatly reduced the invasion ability of Ishikawa cells (P < 0.01). Animal experiments demonstrated that IGSF1 knockdown reduced the number of pulmonary metastatic foci (P < 0.001). On the other hand, IGSF1 overexpression increased Ishikawa cells’ ability to migrate and invade (P < 0.01). IGSF1 overexpression also inhibited E-cadherin expression and promoted that of vimentin (P < 0.001). The expression of c-Myc decreased following IGSF1 knockdown and increased after its overexpression. Silencing of c-Myc reversed the oncogenic effects of IGSF1 (P < 0.01).
Conclusion:
IGSF1 promotes EMT and metastasis in EC through the upregulation of the c-Myc expression. IGSF1 may serve as a potential therapeutic target for EC, and its inhibition can offer new strategies for mitigating the aggressiveness and metastatic potential of this malignancy.
Keywords
Endometrial cancer
Immunoglobulin superfamily member 1
Myc proto-oncogene
Metastasis

INTRODUCTION
Endometrial cancer (EC) belongs to the most common gynecological cancers globally.[1,2] The prognosis for advanced or recurrent EC remains subpar despite the major advancements in the early identification and therapy.[3] The key factors affecting patient prognosis include the invasiveness and metastatic nature of EC.[4,5] Epithelial–mesenchymal transition (EMT) renders tumor cells with invasive and metastatic capabilities, and it involves the transformation of cell phenotype and regulation of multiple signaling pathways.[6] Nevertheless, the precise molecular mechanisms remain unclear; this issue especially holds in EC, where the intricate regulatory network of EMT involves numerous components and pathways.[7] The interactions of these factors and their specific roles in disease progression require further investigation.
Immunoglobulin superfamily member 1 (IGSF1) and the transcription factor Myc proto-oncogene (c-Myc) have garnered widespread attention for their contributions to various cancers.[8,9] The transmembrane protein IGSF1 is aberrantly expressed in some cancers and shows an intimate linkage to the growth and spread of tumors.[10-12] The critical transcription factor c-Myc controls cell division, metabolism, and death. c-Myc also performs a particularly important function in cancer.[13,14] Notably, greater invasiveness and aggressiveness in several cancers have been associated with overexpression of c-Myc.[15-17] Although the previous studies have suggested that IGSF1 and c-Myc may promote tumor invasion and metastasis by influencing EMT in other cancer types, whether this mechanism exists in EC and is the manner of interaction of these factors which remain unclear.
Despite existing research revealing the potential roles of IGSF1 and c-Myc in tumor development, studies on their specific functions and interactions in EC remain very limited.[18,19] At present, no study has defined the detailed molecular mechanisms by which IGSF1 regulates c-Myc and how this process affects EMT and metastasis in Ishikawa cells have not been clearly defined. In addition, systematic studies on the expression patterns and functional effects of these molecules across different molecular subtypes and clinical stages of EC are lacking.
This work aimed to investigate the mechanism underlying IGSF1’s regulation of c-Myc and its roles in EC metastasis and EMT. In vivo animal models and in vitro cellular investigations were used to thoroughly analyze the expression patterns of IGSF1 and c-Myc, explore their interactions in the progression of EC, and examine their effect on EMT and their metastatic capabilities. Furthermore, we investigated whether silencing c-Myc can reverse the malignant behavior promoted by IGSF1 overexpression. This study presents prospective molecular targets for the development of novel therapeutic techniques for EC and offers a fresh viewpoint on the disease’s molecular causes through a thorough exploration of the function and connections of IGSF1.
MATERIAL AND METHODS
Cell culture
The Ishikawa cell line (BFN60805245) was purchased from American Type Culture Collection, Manassas, VA, USA, suspended in complete Dulbecco’s Modified Eagle Medium (DMEM) (BL301A, Biosharp, Anhui, China), and then kept at 37°C with 5% carbon dioxide (CO2) in a humidified chamber. Short tandem repeat profiling confirmed cell authenticity, and mycoplasma testing yielded negative results. Fetal bovine serum, (BL201A, Biosharp, Anhui, China), DMEM, and penicillin/streptomycin (BL505A, Biosharp, Anhui, China) were purchased from Gibco (Rockville, MD, USA). Cell viability was observed using an inverted microscope (IX83P2ZF, Olympus Corporation, Tokyo, Japan), and once, the cell concentration reached 90%, the cells were inoculated on a 6-well plate for 24 h.
Cell transfection
The cultivated Ishikawa cells were diluted before they were plated in six-wells plates, with 1.0 × 105 cells per well. Cell transfection was performed using Lipofectamine 2000 (11668030, Invitrogen, Carlsbad, CA, USA). NC small interfering (siRNA) and IGSF1 siRNA (Yeasen, Shanghai, China) were transfected to Ishikawa cells to create the si-NC (negative control to IGSF1 siRNA) and si-IGSF1 groups, respectively. IGSF1-overexpressing lentivirus (OE-IGSF1) and negative control to IGSF1-overexpressing lentivirus (OENC) and c-Myc interfering lentivirus (sh-c-Myc) and negative control to c-Myc interfering lentivirus (NC-c-Myc) were provided by GenePharma Co., Ltd (Shanghai, China).
c-Myc short hairpin RNA (shRNA): 5’-CAGTTGAAACA CAAACTTGAA-3’
IGSF1 siRNA1 (si-IGSF1-1): Primer forward 5’-CCACCAUGCUGAAGACAUUTT-3’, reversed 5’-AAU GUCUUCAGCAUGGUGGTT-3’
IGSF1 siRNA2 (si-IGSF1-2): Primer forward 5’-GCAUCUAUAGCUGCCACUATT-3’, reversed 5’-UAG UGGCAGCUAUAGAUGCTT-3’
OE-IGSF1: Primer forward 5’-GGGTGACTG GTAAGGTTCTG-3’, reversed 5’-CAACAGGCCCTGT GGTATATC-3’
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using Trizol reagent (R1100, Solarbio, Beijing, China) and reverse transcribed into complementary DNA (cDNA) using the ABScript II cDNA First-Strand Synthesis Kit (RK20400, ABClonal, Wuhan, Hunan, China). Applied Biosystems’ 7500 Real-Time PCR System (7500, Thermo Fisher Scientific, Waltham, MA, USA) and SYBR Green PCR Master Mix (4312704, Thermo Fisher Scientific, Waltham, MA, USA) were used in qRT-PCR analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control to determine the relative expression levels of the following sequences through the 2−ΔΔCT technique.
IGSF1: Primer forward 5'-CGGAAGAGACCAGACC AT-3', reversed 5'-TGATTAGAGACCAAGGAACAG-3'
c-Myc: Primer forward 5'-CGTCTCCACACATCAGC ACAA-3’, reversed 5'-TCTTGGCAGCAGGATAGTCC TT-3'
E-cadherin: Primer forward 5'-ATGCCATCGTTGTT CACTGGA-3', reversed 5'-CATGAGAAGTATGACAA CAGCCT-3'
Vimentin: Primer forward 5'-CCAGGCAAAGCA GGAGTC-3', reversed 5'-GGGTATCAACCAGAGG GAGT-3'
GAPDH: Primer forward 5'-GGTTGAGCAGGTA CTTTT-3', reversed 5'- AGCAAGAGCACAAAG AGGAAG-3'
Western blot
Differently treated cells were collected and lysed in protein lysis buffer (P0013B, Beyotime Biotechnology, Shanghai, China). The protein concentration was measured using a bicinchoninic acid Protein Assay Kit (23227, Pierce, Waltham, MA, USA). After the samples were mixed with the loading buffer, they were heated for 10 min at 95°C. Afterward, the denatured proteins were separated through 15% sodium dodecyl-sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (FFP39, Beyotime Biotechnology, Shanghai, China). The membrane was obstructed using 5% skim milk in bovine serum albumin (BSA, BS114, Biosharp Life Science, Hefei, Anhui, China) for 1 h, then left to incubate overnight at 4°C with IGSF1 primary antibody (PA5-42088, Thermo Fisher Scientific, Waltham, MA, USA1:1000 dilution), c-Myc primary antibody (AF6054, Affinity, Jiangsu, China, 1:1000 dilution), and GAPDH (AF7021, Affinity, Jiangsu, China, 1:3000 dilution). The goat anti-rabbit immunoglobulin G (IgG) linked with horseradish peroxidase (HRP) (#S0001, Affinity, Jiangsu, China, 1:5000 dilution) was left at room temperature for 1 h. Proteins were observed using the Image Quant LAS4000 (GE Healthcare, Chicago, IL, USA) and an enhanced chemiluminescence detection kit (A: B = 1:1, E423-01, Novozan Biotech, Nanjing, China). ImageJ software (v1.8, National Institutes of Health, Bethesda, MD, USA) was utilized to classify the gray values of protein bands, and statistical data were obtained through the normalization method.
Scratch assay for cell migration
Ishikawa cells were cultured in a 6-well plate during their exponential growth phase until 90% confluency. A clean 200 mL pipette tip was utilized to create a scratch on the surface. Following three washes with phosphate buffer saline (PBS), serum-free medium was introduced to the plates. To find the rate of cell migration, we measured the breadth of the wound with an optical microscope at 0 and 24 h.
Transwell migration and invasion assays
A Transwell chamber (24-well plate, 8 mm pore size, 4395; Corning Incorporated, Corning, NY, USA) was used for the migration and invasion assay. To conduct the invasion test, we covered the top chamber with 20 mL 40% Matrigel (356234, Corning Incorporated, Corning, NY, USA) and left it to solidify at 37°C for 15 min. Trypsin was used to treat the EC cells (1 × 105 cells/mL), which were subsequently transferred to the upper chamber (200 mL/well) in serum-free media. The lower chamber was filled with 500 mL complete medium. The plate was incubated at 37°C with 5% CO2 for 48 h. The interior bottom of the upper chamber’s non-migratory cells were cleaned and washed twice with PBS. The cells were fixed for 20 min with 5% paraformaldehyde and then stained for 10 min with 0.4% crystal violet. The cells in 10 randomly selected areas were observed and tallied under an inverted microscope (CKX31, Olympus, Corporation, Tokyo, Japan) at a ×200 magnification.
Immunofluorescence staining
The processed Ishikawa cells were rinsed with PBS and then treated with 4% paraformaldehyde (P1110 Solarbio, Beijing, China) for 30 min at room temperature. Then, they were permeabilized for 15 min with 0.5% Triton X-100 (P0096, Beyotime Biotechnology, Shanghai, China) and cleaned with PBS. After blocking for 30 min at ambient temperature using 10% goat serum, the primary antibodies of IGSF1 (DF14619, Affinity, JiangSu, China, 1:100 dilution) E-cadherin (ab40772, Abcam, Cambridge, MA, USA), vimentin (ab92547, Abcam, Cambridge, MA, USA), and c-Myc (ab78318, Abcam, Cambridge, MA, USA, 1: 100 dilution) were incubated overnight at 4°C. The following day, after the removal of the primary antibody, goat anti-rabbit IgG H&L (Alexa Fluor® 488 and Alexa Fluor® 594) was added, and the resulting mixture was incubated in the dark and at room temperature for 1 h. The secondary antibodies (ab150077 and ab150080) were obtained from Abcam (Cambridge, MA, USA). Then, the secondary antibodies were removed. The cells were then exposed to 4’,6-diamidino-2-phenylindole (DAPI, C1002, Beyotime Biotechnology, Shanghai, China) for 10 min at room temperature before the final PBS wash. A Leica Microsystems fluorescence microscope (M125, Wetzlar, Germany) was used to capture images.
Lung metastasis animal model
A total of 42 female BALB/c nude mice, between the ages of 4 and 6 weeks, were acquired from Beijing Vital River Laboratory Animal Technology Co., Ltd. (license number SCXK [Jing] 2016-0006) and placed in the specific pathogen free-level laboratory at the Animal Experimentation Center. This study was approved by the ethics committee of Qingdao Municipal Hospital (Group), with the approval No. 20221323. The mice were randomly divided into seven groups, namely, NC, shNC, sh-IGSF1, OE-NC, OE-IGSF1, OE-IGSF1 + NC-c-Myc, and OE-IGSF1 + sh-c-Myc, with each group having six mice. ShRNA technology was used in animal studies to decrease the expression of IGSF1 in the Ishikawa cell line, which resulted in the establishment of stable IGSF1 knockdown (sh-IGSF1) and negative control to stable IGSF1 knockdown (sh-NC) cell lines. The mice were intravenously injected through the tail vein with 0.2 mL cell suspension, with each dose containing 5 × 106 cells/200 mL. The general condition of the mice (mental state, activity, diet, and body weight) was observed every 2–3 days. After 6 weeks of inoculation, the mice were sacrificed through neck dislocation, and their lung tissues were collected for histological analysis to assess tumor development. Hematoxylin and eosin (H&E) staining (C0105S, Beyotime Biotechnology, Shanghai, China) was performed after the lung tissues were embedded in paraffin, fixed in neutral buffered formalin, and sectioned. Number of lung metastases were examined using an optical microscope (BX46, Olympus, Tokyo, Japan).
sh-IGSF1: 5’-CAAAGAUGGAAGUGAAAUAGCAUCC-3’
Immunohistochemical staining
The related methods were performed referring to the work of Higashine et al.[20] The fixed tissue was dehydrated, embedded in paraffin, and sliced (4 mm). Sections were treated to remove paraffin and rehydrated, blocked after antigen retrieval, and incubated with the primary antibodies of IGSF1 (DF14619, Affinity, JiangSu, China, 1:200 dilution), E-cadherin (ab314063, Abcam, Cambridge, MA, USA, 1:200 dilution), and vimentin (ab45939, Abcam, Cambridge, MA, USA, 1:200 dilution) for an additional night at 4℃. The following day, a secondary antibody goat anti-rabbit IgG H&L (HRP) (ab6721, Abcam, Cambridge, MA, USA) was applied and left to incubate at room temperature for half an hour, followed by a 15 min diaminobenzidine staining process, hematoxylin counterstaining, dehydration, and mounting for microscopic examination. The degree of staining and the proportion of positive cells dictated the results of immunohistochemical staining. Scores for stain intensity are as follows: 0 represents no color, 1 is light yellow, 2 is brown-yellow, and 3 is dark brown. Cell positivity percentages were scored as follows: 0 represents ≤5%, 1 represents 6–25%, 2 represents 26–50%, 3 represents 50–75%, and 4 represents more than 75%. The ultimate immunohistochemical staining score was determined by multiplying the scores for intensity of staining and proportion of positive cells, with scores ranging from 0 to 4 representing negative (−), 5–8 representing positive (+), and 9–12 representing strongly positive (+++).[21-23]
5-ethynyl-2’-deoxyuridine (EdU) assay
The transfected Ishikawa cells were distributed to a 96-well plate, with 1 × 104 cells in each well. Following a 24 h culture period, 20 mmoL/L EdU (Cat: CA1170, Solarbio, Beijing, China) was introduced to each well and allowed to incubate at 37℃ for 2 h. Using 0.5% Triton X-100, the cells were permeabilized following their fixation in 4% paraformaldehyde. Next, they were stained with 100 mL 1X Apollo567 fluorescent dye (Guangzhou RiboBio Co., Ltd., Guangzhou, China) and 100 mL DAPI for 30 min each. EdU-positive cells were identified using Apollo567 fluorescent dye and a fluorescence microscope.
Statistical analysis
The experimental data were analyzed using the Statistical Package for the Social Sciences 20.0 (IBM Corp., Armonk, NY, USA). One-way analysis of variance was used for comparisons among multiple groups, and Student’s t-test was used for comparisons between two groups. Graphs were plotted using GraphPad Prism 7 (Inc., San Diego, CA, USA). Data were shown as the mean ± standard deviation. Statistical significance was determined at P < 0.05, 0.01, and 0.001. The experiments were conducted thrice for accuracy.
RESULTS
Knockdown of IGSF1 inhibited the metastasis of Ishikawa cells
The levels of IGSF1 protein expression in Figure 1a and b were assessed through Western blot analysis. Compared with the negative control (siNC) group, the si-IGSF1-1 and si-IGSF1-2 groups displayed a significant drop in IGSF1 expression, which indicates effective siRNA interference and robust IGSF1 expression suppression (P < 0.001). Subsequently, the migration capabilities of cells post-IGSF1 knockdown were assessed through wound healing assay. Results show that cells in the siNC group had the strongest migration capability, and those in si-IGSF1-1 and si-IGSF1-2 groups exhibited significantly reduced migration to the scratch area, which suggests that knockdown of IGSF1 inhibited the migration ability of Ishikawa cells [Figure 1c and d], (P < 0.001). The siNC group exhibited the greatest number of cells migrating through the Matrigel matrix and holes in the migration and invasion studies. Conversely, silencing of IGSF1 led to a notable decrease in the quantity of cells moving through, which implies a decline in cell migration and invasion abilities due to IGSF1 deficiency [Figure 1e and f], (P < 0.001). The qRT-PCR findings [Figure 1g and h] demonstrate that the reduction in IGSF1 led to high levels of E-cadherin messenger RNA (mRNA) and low levels of vimentin mRNA, which suggests the inhibition of the EMT process (P < 0.01). Furthermore, these findings were confirmed by immunofluorescence staining. In comparison with the siNC group, knock down of IGSF1 enhanced the fluorescence of E-cadherin and decreased the fluorescence of vimentin, which indicates that IGSF1 knockdown reduced EMT [Figure 1i]. In conclusion, the experimental results in Figure 1 reveal that knockdown of IGSF1 can inhibit EMT and reduce the migration and invasion capabilities of Ishikawa cells. This finding implies that IGSF1 may be an important regulatory factor in the metastasis of EC.
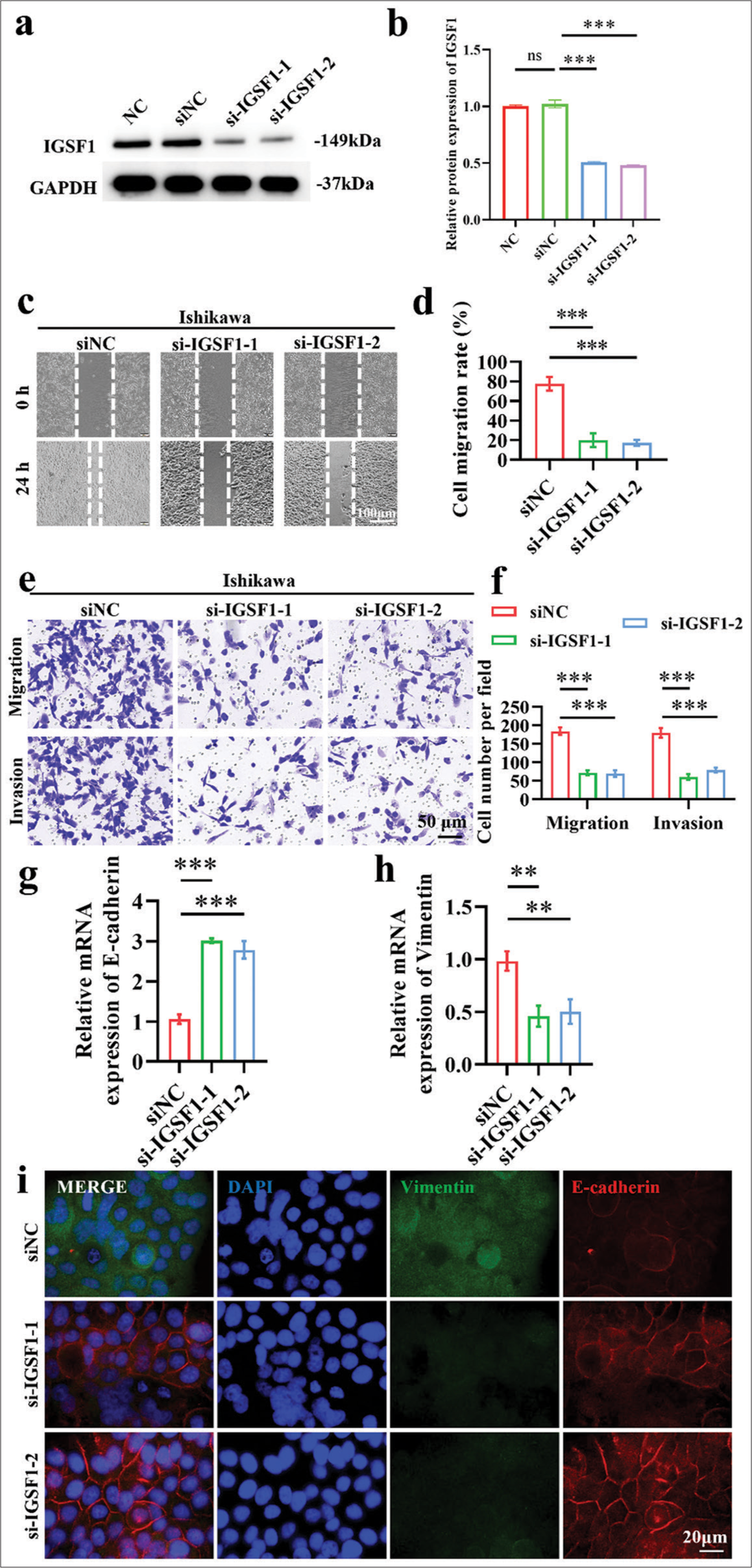
- Knockdown of immunoglobulin superfamily member 1 (IGSF1) inhibited metastatic potential in Ishikawa cells. (a and b) Validation of small interfering RNA-mediated knockdown efficiency of IGSF1. (c and d) Wound healing assay demonstrating that IGSF1 knockdown suppressed the migration of Ishikawa cells. (e and f) IGSF1 knockout Ishikawa cells were analyzed through Transwell assay. (g and h) Quantitative analysis of the changes in the expressions of E-cadherin (g) and vimentin (h) following IGSF1 knockdown. (i) Immunofluorescence dual staining analysis of E-cadherin and vimentin post-IGSF1 knockdown. n = 3, ns: No statistical significance, **P < 0.01, ***P < 0.001. DAPI,4’,6-Diamidino-2’-phenylindole. NC, negative control; si-NC, negative control to IGSF1 siRNA; si-IGSF1, IGSF1 siRNA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Animal experimental results show that the knockdown of IGSF1 inhibited the lung metastasis capability of Ishikawa cells
First, the body weight of sh-NC and sh-IGSF1 groups gradually increased, and the sh-IGSF1 group presented a higher final body weight than the sh-NC [Figure 2a]. Western blot analysis confirmed the significantly lower expression of IGSF1 in sh-IGSF1 cells compared with those in the sh-NC group [Figure 2b and c], (P < 0.001), which indicates the successful construction of the knockdown cell line. Afterward, the sh-NC and sh-IGSF1 groups were administered through the tail veins of nude mice, with evaluation of lung metastasis performed 6 weeks later. The H&E staining results reveal the multiple metastatic foci in the lungs of the sh-NC group, whereas the sh-IGSF1 group exhibited a notable decrease in lung metastatic foci count [Figure 2d and e], (P < 0.001). This finding implies that the suppression of IGSF1 can hinder the lung metastasis ability of Ishikawa cells. To further explore the mechanism, we performed immunohistochemical staining for E-cadherin and vimentin on lung metastatic foci. In the comparison of the lung metastatic foci of the sh-IGSF1 and sh-NC groups, immunohistochemical examination revealed the higher E-cadherin (P < 0.01) expression and lower vimentin (P < 0.05) expression [Figure 2f-h]. These results imply that IGSF1 suppression impedes EMT in cancer cells.
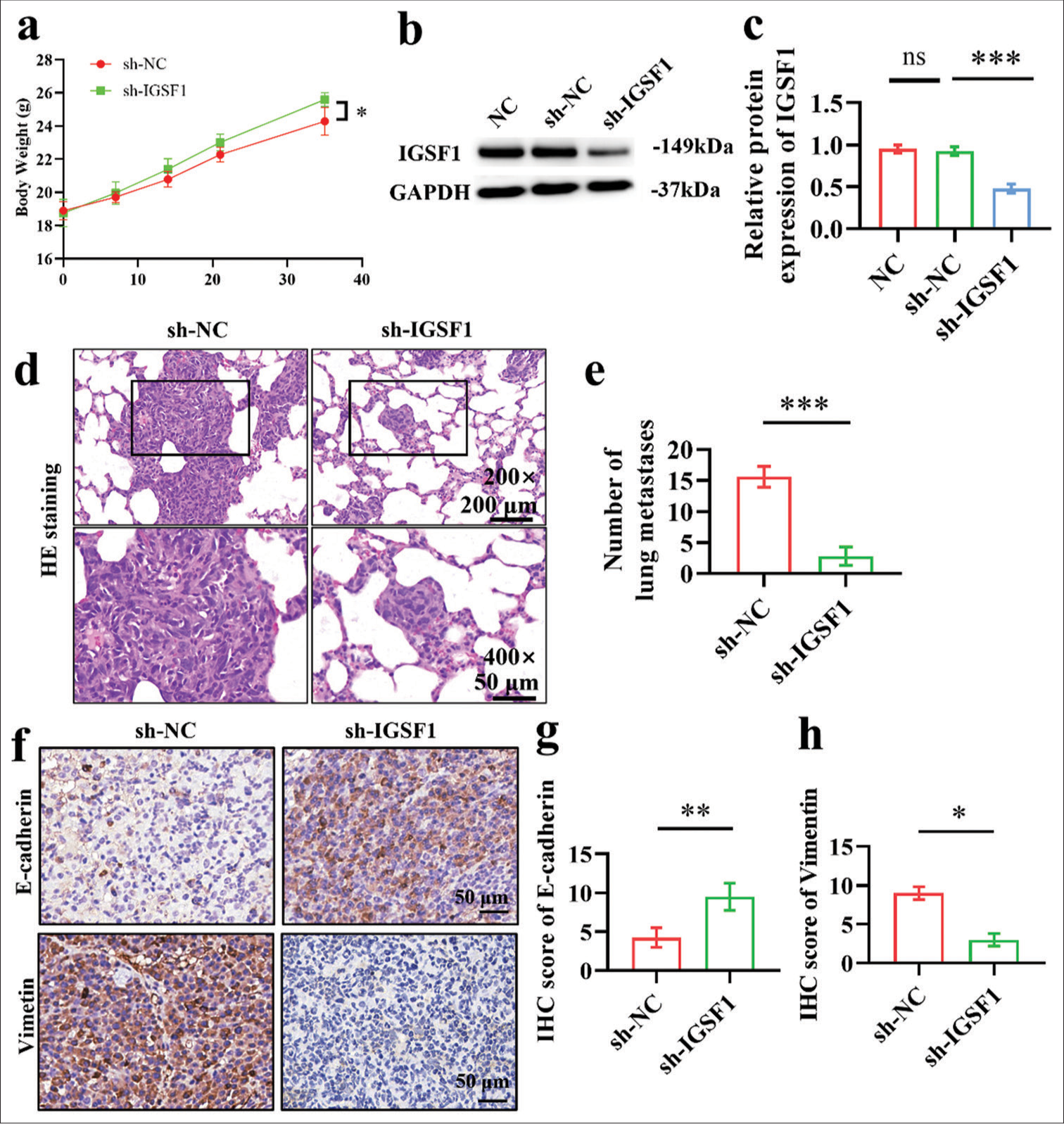
- Animal model results demonstrate that immunoglobulin superfamily member 1 (IGSF1) knockdown inhibited lung metastasis of Ishikawa cells. (a) Body weight of mice with lung metastasis model. (b and c) Verification of IGSF1 knockdown effectiveness in stable Ishikawa cell lines. (d and e). Detection of lung metastases 6 weeks after tail vein injection in nude mice. (Scale: 200 mm, objective: ×200) (scale: 50 mm, objective: ×400). (f-h) Immunohistochemical staining of E-cadherin and vimentin in lung metastatic lesions. Scale bar: 50 mm. n = 6, ns: No statistical significance, *P < 0.05, **P < 0.01, ***P < 0.001. NC, negative control; sh-NC, negative control to stable IGSF1 knowown; sh-IGSF1, stable IGSF1 knowown; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Effect of IGSF1 overexpression on Ishikawa cells
Comparison of the OE-IGSF1 and OE-NC group through Western blot analysis revealed the significantly higher IGSF1 expression in the former [Figure 3a and b], (P < 0.001), which demonstrates the successful establishment of the IGSF1-overexpression cell line. Transwell and scratch tests were conducted to assess the effect of elevated IGSF1 expression on the invasive and migrating characteristics of Ishikawa cells. Scratch test findings indicate a notable increase in the quantity of cells that moved toward the scratch region in the OE-IGSF1 group in contrast to the OE-NC group. This result implies that the heightened IGSF1 expression boosted the migratory capacity of Ishikawa cells [Figure 3c and d], (P < 0.01). In line with this finding, the Transwell assay demonstrated a notable increase in the number of cells that migrated through the chamber membrane in the OE-IGSF1 group compared with the OE-NC group [Figure 3e and f], (P < 0.01), which further proves that elevated levels of IGSF1 can enhance the migratory and invasive properties of Ishikawa cells. [Figure 3g and h] indicated that after IGSF1 overexpression, E-cadherin mRNA levels were significantly downregulated, and vimentin mRNA expression was upregulated (P < 0.001). This finding suggests that IGSF1 may improve the invasive and metastatic capabilities of Ishikawa cells by promoting epithelial–mesenchymal transition (EMT). Finally, 5-ethynyl-2’-deoxyuridine ( EdU) incorporation assay was performed to analyze the effect of IGSF1 overexpression on Ishikawa cell proliferation. The findings indicate that the proportion of EdU-positive cells was notably elevated in the OE-IGSF1 group in contrast to that in the OE-NC group, which suggests that IGSF1 overexpression not only boosted invasion and metastasis but also amplified cell proliferation [Figure 3i and j], (P < 0.01).
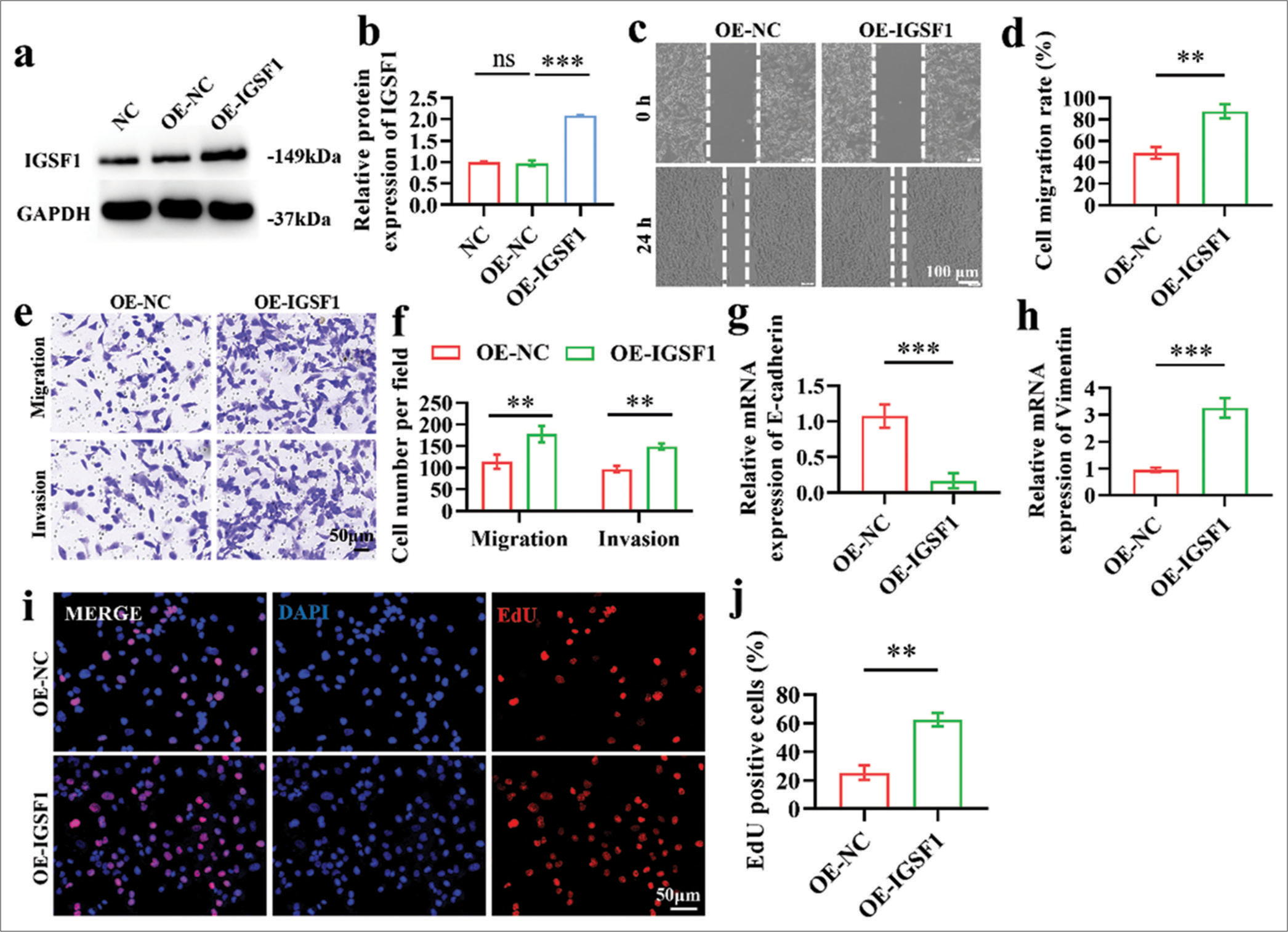
- Effect of immunoglobulin superfamily member 1 (IGSF1) overexpression on Ishikawa cells. (a and b) Verification of overexpression efficiency of OE-IGSF1. (c and d) Wound healing assay indicating the enhanced migration of Ishikawa cells with IGSF1 overexpression. Scale bar: 100 mm. (e and f) Transwell assays showing increased migration and invasion capabilities. Scale bar: 50 mm. (g and h) Quantitative analysis using quantitative real-time polymerase chain reaction conducted to assess the changes in the expression of E-cadherin (g) and vimentin (h) markers following the overexpression of IGSF1. (i and j). 5-ethynyl-2’-deoxyuridine assay illustrating changes in cell the proliferation capacity after IGSF1 overexpression. Scale bar: 50 mm. n = 3, ns: No statistical significance, **P < 0.01, ***P < 0.001. NC, negative control; OE-NC, negative control to IGSF1-overexpressing lentivirus; OE-IGSF1, IGSF1-overexpressing lentivirus; GAPDH, glyceraldehyde-3-phosphate dehydrogenase .
c-Myc expression was increased by IGSF1
The si-IGSF1-1 and si-IGSF1-2 groups exhibited significantly lower c-Myc mRNA expression levels than the siNC group following IGSF1 silencing, and this result suggests that IGSF1 silencing can inhibit c-Myc transcription [Figure 4a], (P < 0.001). The findings indicate that the c-Myc protein expression levels were notably decreased in the IGSF1 silenced groups (si-IGSF1-1 and si-IGSF1-2) in comparison with the siNC group, which suggests that the suppression of IGSF1 can impede the translation level of c-Myc [Figure 4b and c], (P < 0.001). [Figures 4d-f] indicate that the upregulation of IGSF1 led to a notable increase in c-Myc mRNA and protein levels compared with those in the OE-NC group. The results imply that the overexpression of IGSF1 enhanced the transcription and translation of c-Myc (P < 0.001). Immunofluorescence double staining can visually display the expression localization of proteins. [Figure 4g] demonstrates the notable increase in the fluorescence intensity of c-Myc protein in the OE-IGSF1 group compared with that in the OE-NC group. This finding suggests that IGSF1 overexpression can increase the expression of c-Myc protein. In general, c-Myc mRNA and protein production can be inhibited by decreasing IGSF1, while c-Myc mRNA and protein production can be increased by elevating IGSF1 levels. These results suggest that IGSF1 can positively regulate the expression of c-Myc and provide important insights into further studies on the molecular mechanisms by which IGSF1 promotes EC progression.
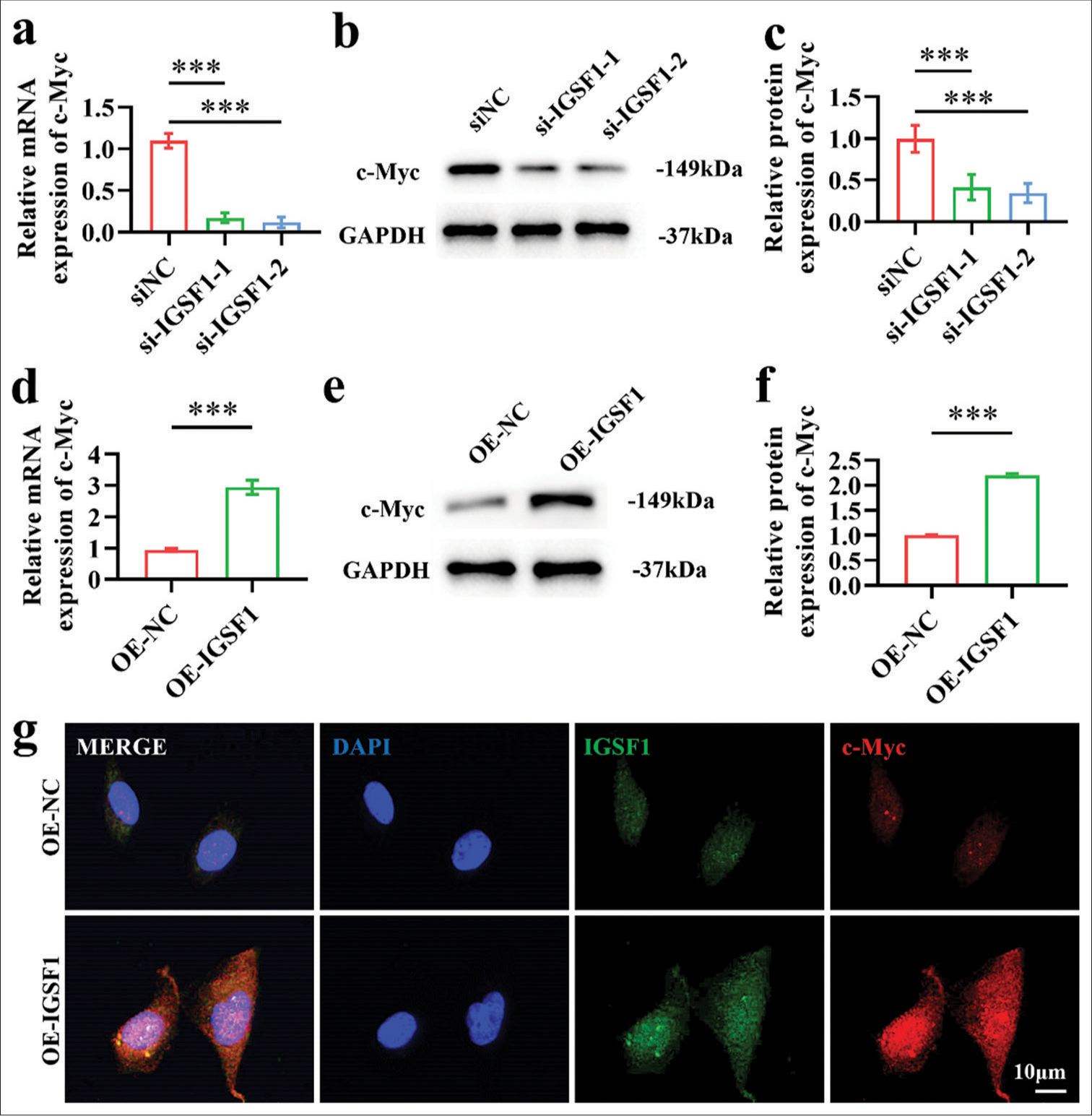
- Myc proto-oncogene (c-Myc) expression was increased by immunoglobulin superfamily member 1 (IGSF1). (a-c) Confirmation of c-Myc expression after IGSF1 knockdown. (d-f) Verification of c-Myc expression after IGSF1 overexpression. (g) Immunofluorescence dual staining. Scale bar: 10 mm. n = 3, ns: No statistical significance. ***P < 0.001. siNC, negative control to IGSF1 siRNA; si-IGSF1, IGSF1 siRNA; OE-NC, negative control to IGSF1-overexpressing lentivirus; OE-IGSF1, IGSF1-overexpressing lentivirus; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Silencing c-Myc reverses the oncogenic effects of IGSF1
We further explored the role of c-Myc in the progression of EC promoted by IGSF1. Figures 5a and b show the association of elevated IGSF1 expression with the significant rise in IGSF1 and c-Myc mRNA levels (P < 0.001). Interestingly, silencing c-Myc did not affect the expression of IGSF1 [Figure 5a], (P > 0.05), which suggests that c-Myc may be a downstream effector molecule of IGSF1. To validate this hypothesis, we silenced c-Myc in IGSF1-overexpressing cells and assessed the influence on IGSF1’s oncogenic effects through a series of functional experiments. Transwell migration and invasion tests revealed that elevated levels of IGSF1 notably boosted the invasiveness of EC cells. The OE-NC group exhibited a significantly lower value than the OE-IGSF1 group, and the OE-IGSF1+sh-c-Myc group attained a significantly lower result than the OE-IGSF1+NC-c-Myc group (P < 0.001) [Figure 5c-e]. Silencing of c-Myc in the context of IGSF1 overexpression reduced the number of invading cells to 48.333 ± 4.714 (P < 0.001), which suggests a key role for c-Myc in mediating the pro-invasive effects of IGSF1. EdU incorporation assay was performed to further examine the proliferative capacity of cells in each group. [Figures 5f and g] demonstrate that the proliferation of EC cells significantly increased with IGSF1 overexpression, as evidenced by the increase in EdU-positive cells from 29.795% ± 7.86% to 62.037% ± 4.083% in the control group (P < 0.001). After the silencing of c-Myc, the proportion of EdU-positive cells dropped to 20.728% ± 3.653% (P < 0.001), which indicates the crucial role of c-Myc in enabling IGSF1 to promote cell proliferation. The OE-IGSF1 + sh-c-Myc group exhibited a substantially lower cell mobility than the OE-IGSF1 + NCc-Myc group, according to the results of cell scratch assay (P < 0.001) [Figure 5h-i]. These findings imply that IGSF1 upregulates c-Myc to cause EMT, which, in turn, encourages EC cells to invade and spread. Finally, by examining the changes in the expression of E-cadherin and vimentin, which are indicators of EMT, we were able to determine how IGSF1 controls the progression of EC through the regulation of EMT. The qRT-PCR findings indicate that the upregulation of IGSF1 resulted in a 0.4-fold reduction in the epithelial indicator E-cadherin and a 3-fold rise in the mesenchymal indicator vimentin, which suggests that IGSF1 facilitates EMT [Figure 5j and k]. The OE-IGSF1 + sh-c-Myc group displayed a large increase in E-cadherin expression compared with the OE-IGSF1 group but a significant decrease in vimentin expression (P < 0.001).
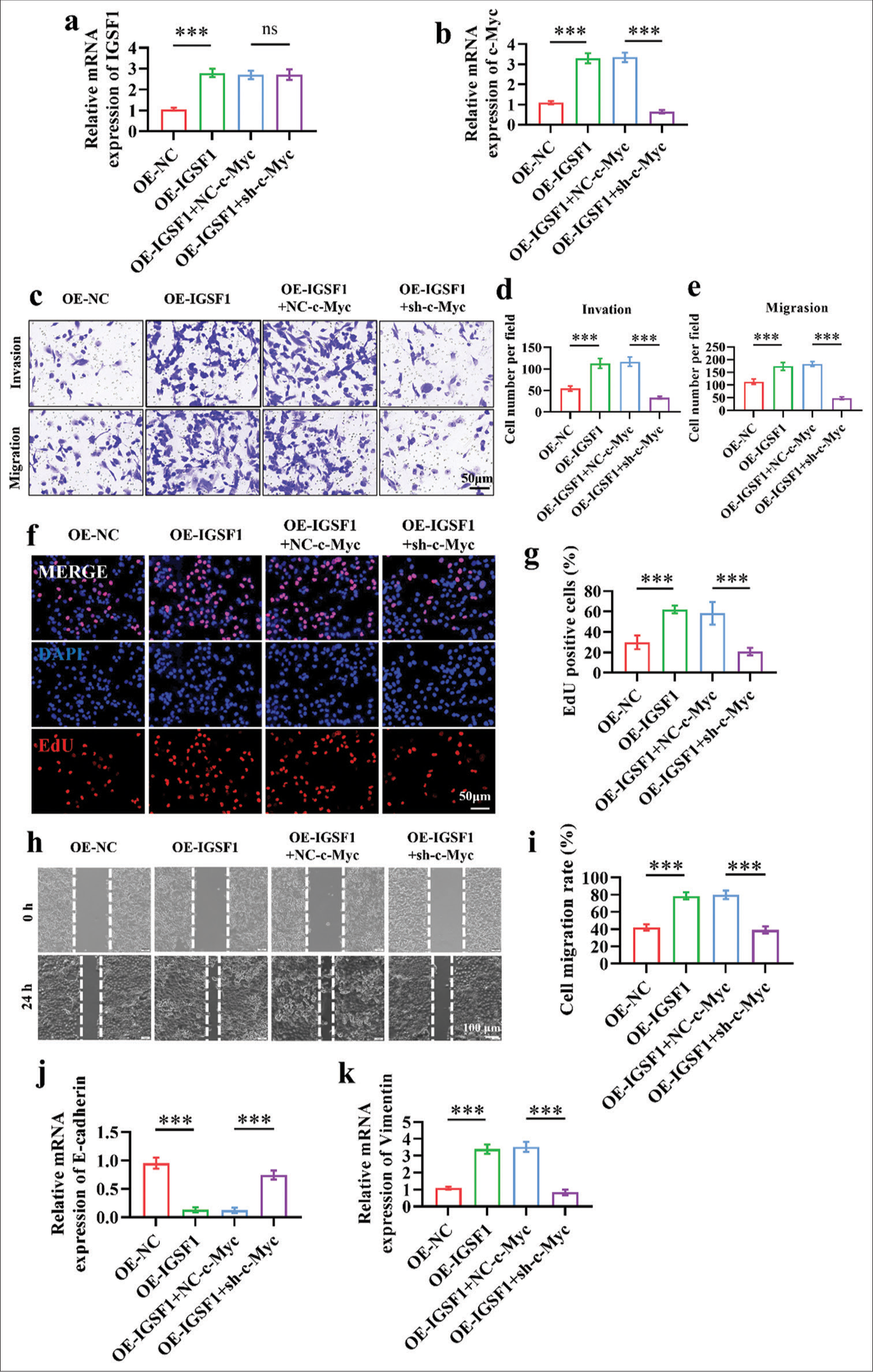
- Silencing of Myc proto-oncogene (c-Myc) reversed the oncogenic effects of immunoglobulin superfamily member 1 (IGSF1). (a and b) Quantitative real-time polymerase chain reaction verification of IGSF1 expression (a) and c-Myc (b) changes in different treatment groups. (c-e) Transwell assay conducted to assess the effect of c-Myc silencing on the oncogenic effects of IGSF1. Scale bar: 50 mm. (f and g) 5-ethynyl-2’-deoxyuridine assay for the evaluation of the influence of c-Myc silencing on the oncogenic effects of IGSF1. Scale bar: 50 mm. (h and i) Wound healing assay performed to evaluate the influence of c-Myc silencing on the oncogenic effects of IGSF1. Scale bar: 100 mm. (j and k) Detection of E-cadherin (j) and vimentin (k) expression. n = 3, ns: No statistical significance, ***P < 0.001. OE-NC, negative control to IGSF1-overexpressing lentivirus; OEIGSF1, IGSF1-overexpressing lentivirus; NC-c-Myc, negative control to c-Myc interfering lentivirus; sh-Myc, c-Myc interfering lentivirus.
Silencing of c-Myc reversed the pro-metastatic effects of IGSF1 in the lung
In comparison with the OE-NC and OE-IGSF1 + sh-c-Myc groups, the OE-IGSF1 and OE-IGSF1 + NC-c-Myc groups showed a notably higher number of lung metastatic foci, which indicates that IGSF1 overexpression may enhance lung metastasis, whereas inhibition of c-Myc downstream can counteract this effect [Figure 6a and b], (P < 0.01). In addition, the mice in the OE-IGSF1 + sh-c-Myc group had a higher ultimate body weight than the animals in the OE-IGSF1 + NC-c-Myc group [Figure 6c]. E-Cadherin expression levels in lung metastatic foci were notably increased in the OE-NC and OE-IGSF1 + sh-c-Myc groups compared with those in the OE-IGSF1 and OE-IGSF1 +NC-c-Myc groups [Figure 6d], (P < 0.01). IGSF1 overexpression suppressed the expression of E-cadherin and induced EMT, and c-Myc silencing blocks this effect. Vimentin expression levels in lung metastatic foci were notably elevated in the OE-IGSF1 and OE-IGSF1+NC-c-Myc groups compared with those in the OE-NC and OE-IGSF1 + sh-c-Myc groups [Figure 6e], (P < 0.001). Overexpression also promoted EMT, and this result further verifies that IGSF1 overexpression can induce EMT, and silencing of c-Myc can block the EMT induced by IGSF1.
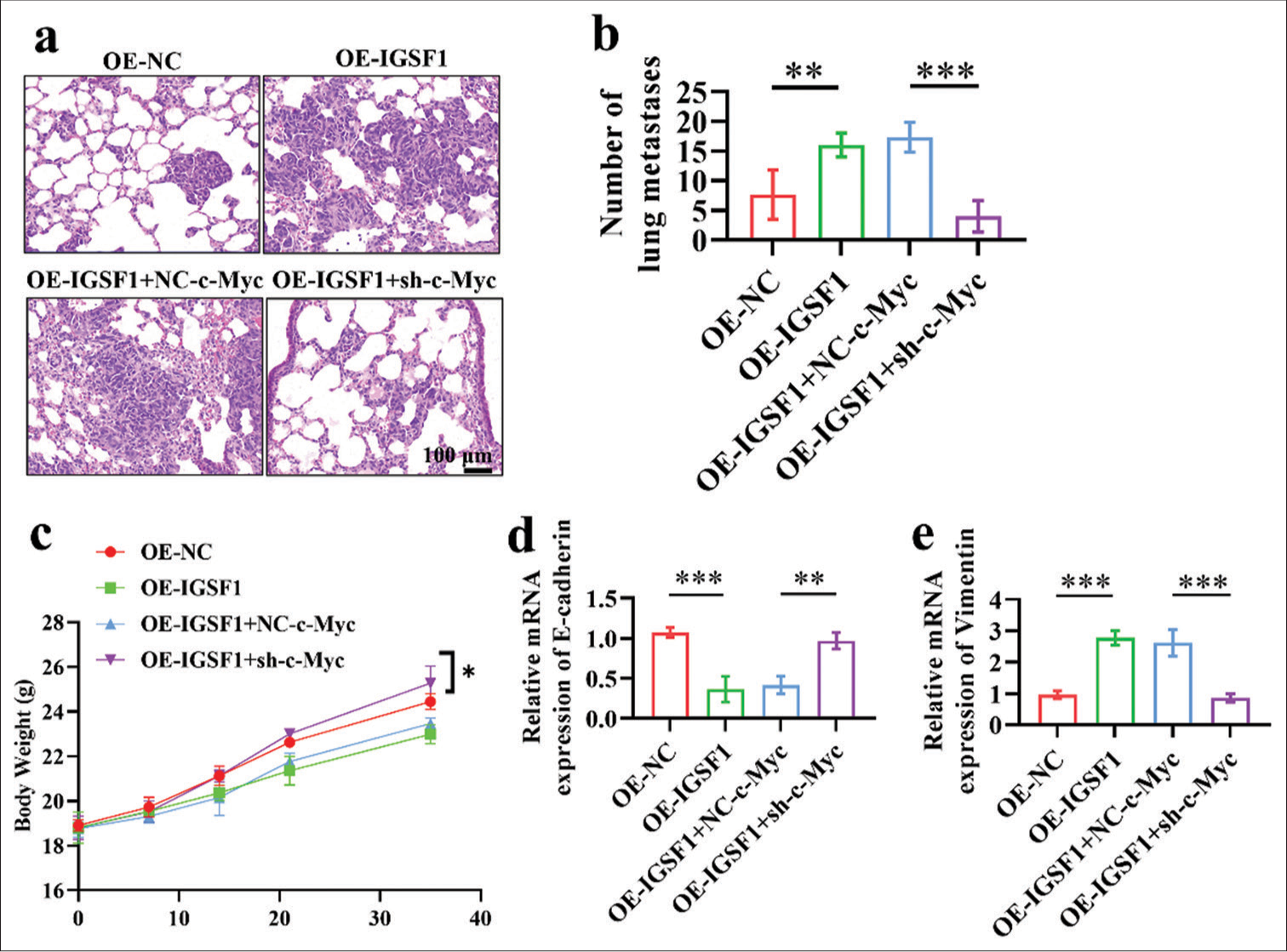
- Silencing of Myc proto-oncogene reversed the prometastatic effects of immunoglobulin superfamily member 1 on lung metastasis. (a and b) Detection of lung metastases 6 weeks after tail vein injection of cells in nude mice. Scale bar: 100 mm. (c) The mice body weight. (d and e) E-cadherin (d) and vimentin (e) in lung metastatic lesions. n = 6, *P < 0.05, **P < 0.01, ***P < 0.001. OE-NC, negative control to IGSF1-overexpressing lentivirus; OE-IGSF1, IGSF1-overexpressing lentivirus; NC-c-Myc, negative control to c-Myc interfering lentivirus; sh-Myc, c-Myc interfering lentivirus.
DISCUSSION
IGSF1 exhibits an aberrant expression in various malignancies and linked to the invasiveness and potential for tumor metastasis.[8,10] Combined with our results, the do-regulation of IGSF1 expression inhibited cell activity and invasion in EC cells. In addition, as a key transcription factor, the role of c-Myc in tumor development has been extensively studied.[24-26] Studies also revealed a correlation between elevated tumor aggressiveness and c-Myc overexpression.[27-29] Our research further clarified the roles of IGSF1 and c-Myc in EC, particularly their interactions during the EMT process; the discovered new molecular mechanisms provide additional insights into the metastasis of EC.[30] We discovered that IGSF1 knockdown strongly suppressed the migratory and invasive potential of Ishikawa cells and the suppressed EMT process. Conversely, the overexpression of IGSF1 improved the migratory and invasive capabilities of these cells and promoted the EMT process. Finally, we confirmed the mediating role of c-Myc in IGSF1-regulated EMT and metastasis in Ishikawa cells, where the silencing of c-Myc reversed the enhanced effects of EMT and metastasis caused by IGSF1 overexpression. Moreover, we validated the effect of IGSF1 on EC metastasis through animal experiments. First, the overexpression of IGSF1 reduced the body weight of mice, and after the silencing of c-Myc, the body weight of mice gradually increased and approached that of control mice. Knockdown of IGSF1 significantly reduced lung metastatic foci in a nude mouse model, and this result further confirms the critical role of IGSF1 in the invasiveness and metastatic capabilities of EC. These findings correspond with earlier research on the function of IGSF1 in other tumor types, which discovered a correlation between IGSF1 expression and the aggressiveness of other cancers. Previous studies have discussed the regulation of miRNA on downstream genes through IGSF1, CUE domain-containing protein 1, and leucine-rich repeat-containing G protein-coupled receptor 4 (LGR4). The findings indicate that miR-34c-5p affected LGR4 but not IGSF1. Additional experimental findings demonstrate that Circ 0003945 silencing inhibited the representative downstream target genes of the b-catenin pathway (c-Myc), with miR-34c-5p contributing to this process.[31] Combined with our experimental results, it is speculated that IGSF1 and c-Myc may also be related through the bcatenin pathway.
This study clarified the function of IGSF1 in EC, particularly its key role in EMT regulation and promotion of tumor metastasis. This study unveiled the mediating role of c-Myc as a downstream effector molecule of IGSF1 in EMT and EC metastasis and provides a theoretical basis for targeted therapy against c-Myc. We enhanced the scientific validity and applicability of this discovery through experimental verification of the interaction between IGSF1 and c-Myc. These achievements not only enrich the pathophysiological knowledge on EC but may also provide targets for the development of new therapeutic strategies.
Notwithstanding the accomplishments of this research, certain restrictions remain. This research primarily relied on the use of Ishikawa cells. Future studies should validate the role of IGSF1 in a broad range of EC cells and clinical samples. This work mainly focused on the role of IGSF1 and c-Myc in EMT. Their functions in other biological processes in EC, such as cell proliferation and death, should be investigated in the future. Finally, given the diverse biological functions of IGSF1 and c-Myc, future research should further explore their network regulatory mechanisms in EC and determine how to utilize this knowledge to develop more effective treatment strategies.
SUMMARY
Our research discovered that IGSF1 increased the production of c-Myc, which, in turn, increased the growth and invasiveness metastasis of EC cells. Silencing of c-Myc can effectively reverse the oncogenic effects of IGSF1. The study also identified a novel IGSF1/c-Myc/EMT signaling pathway that contributes to the development of EC. These findings provide fresh experimental support for using IGSF1 and c-Myc as therapeutic targets in EC.
AVAILABILITY OF DATA AND MATERIALS
Data to support the findings of this study are available on reasonable request from the corresponding author.
ABBREVIATIONS
IGSF1 – Immunoglobulin superfamily member 1
EC – Endometrial cancer
EMT – Epithelium mesenchymal transition
c-Myc – Myc proto-oncogene
EdU – 5-ethynyl-2’-deoxyuridine
H&E – Hematoxylin and eosin
SPF – Specific pathogen free
GAPDH – Glyceraldehyde-3-phosphate dehydrogenase
ECL – Enhanced chemiluminescence
HRP – Horseradish peroxidase
qRT-PCR – Quantitative real-time polymerase chain reaction
AUTHOR CONTRIBUTIONS
JW, SZ, and JJ: Designed the research study; SD and JJ: Performed the research; JW and SZ: Collected and analyzed the data. All authors have been involved in drafting the manuscript and all authors have been involved in revising it critically for important intellectual content. All authors give final approval of the version to be published. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study has been approved by the ethics committee of Qingdao Municipal Hospital (Group), approval No. 20221323.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
References
- Endometrial cancer In: Cancer screening. CRC Press; 2021. p. :531-56.
- [CrossRef] [PubMed] [Google Scholar]
- Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med. 2023;388:2145-58.
- [CrossRef] [PubMed] [Google Scholar]
- An EMT-related genes signature as a prognostic biomarker for patients with endometrial cancer. BMC Cancer. 2023;23:879.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular landscape of the epithelial-mesenchymal transition in endometrioid endometrial cancer. J Clin Med. 2021;10:1520.
- [CrossRef] [PubMed] [Google Scholar]
- UBE2C is upregulated by estrogen and promotes epithelial-mesenchymal transition via p53 in endometrial cancer. Mol Cancer Res. 2020;18:204-15.
- [CrossRef] [PubMed] [Google Scholar]
- Expression profile of epithelial-mesenchymal transition-related genes as a prognostic biomarker for endometrial cancer. J Cancer. 2021;12:6484.
- [CrossRef] [PubMed] [Google Scholar]
- IGSF1: A novel oncogene regulates the thyroid cancer progression. Cell Biochem Funct. 2019;37:516-24.
- [CrossRef] [PubMed] [Google Scholar]
- The immune suppressor IGSF1 as a potential target for cancer immunotherapy. Cancer Immunol Res. 2024;12:491-507.
- [CrossRef] [PubMed] [Google Scholar]
- The IGSF1, Wnt5a, FGF14, and ITPR1 gene expression and prognosis hallmark of prostate cancer. Rep Biochem Mol Biol. 2022;11:44-53.
- [CrossRef] [PubMed] [Google Scholar]
- From consternation to revelation: Discovery of a role for IGSF1 in pituitary control of thyroid function. J Endocr Soc. 2018;2:220-31.
- [CrossRef] [PubMed] [Google Scholar]
- IGSF1 deficiency results in human and murine somatotrope neurosecretory hyperfunction. J Clin Endocrinol Metab. 2020;105:e70-84.
- [CrossRef] [PubMed] [Google Scholar]
- MYC as a target for cancer treatment. Cancer Treat Rev. 2021;94:102154.
- [CrossRef] [PubMed] [Google Scholar]
- Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol Cancer. 2021;20:3.
- [CrossRef] [PubMed] [Google Scholar]
- C-myc contributes to malignancy of lung cancer: A potential anticancer drug target. Anticancer Res. 2020;40:609-18.
- [CrossRef] [PubMed] [Google Scholar]
- c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat Commun. 2020;11:4980.
- [CrossRef] [PubMed] [Google Scholar]
- Is IGSF1 involved in human pituitary tumor formation? Endocr Relat Cancer. 2015;22:47-54.
- [CrossRef] [PubMed] [Google Scholar]
- IGSF1 mutations are the most frequent genetic aetiology of thyrotropin deficiency. Eur J Endocrinol. 2022;187:787-95.
- [CrossRef] [PubMed] [Google Scholar]
- Promotion of differentiation in developing mouse cerebellar granule cells by a cell adhesion molecule BTIgSF. Neurosci Lett. 2018;686:87-93.
- [CrossRef] [PubMed] [Google Scholar]
- Immunohistochemistry scoring of breast tumor tissue microarrays: A comparison study across three software applications. J Pathol Inform. 2022;13:100118.
- [CrossRef] [PubMed] [Google Scholar]
- Serous ovarian tumors: Immunohistochemical profiling as an aid to grading and understanding tumorigenesis. Med J Armed Forces India. 2020;76:30-6.
- [CrossRef] [PubMed] [Google Scholar]
- Hypoxia-induced vasculogenic mimicry formation in human colorectal cancer cells: Involvement of HIF-1a, Claudin-4, and E-cadherin and Vimentin. Sci Rep. 2016;6:37534.
- [CrossRef] [PubMed] [Google Scholar]
- C-myc amplification in breast cancer: A meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83:1688-95.
- [CrossRef] [PubMed] [Google Scholar]
- The impact of C-MYC gene expression on gastric cancer cell. Mol Cell Biochem. 2010;344:125-35.
- [CrossRef] [PubMed] [Google Scholar]
- Function of the c-Myc oncogenic transcription factor. Exp Cell Res. 1999;253:63-77.
- [CrossRef] [PubMed] [Google Scholar]
- A novel hypoxic long noncoding RNA KB-1980E6. 3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40:1609-27.
- [CrossRef] [PubMed] [Google Scholar]
- NCAPD3 enhances Warburg effect through c-myc and E2F1 and promotes the occurrence and progression of colorectal cancer. J Exp Clin Cancer Res. 2022;41:198.
- [CrossRef] [PubMed] [Google Scholar]
- PRMT5-dependent transcriptional repression of c-Myc target genes promotes gastric cancer progression. Theranostics. 2020;10:4437.
- [CrossRef] [PubMed] [Google Scholar]
- Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch Toxicol. 2021;95:2279-97.
- [CrossRef] [PubMed] [Google Scholar]
- Hsa_circ_0003945 promotes progression of hepatocellular carcinoma by mediating miR-34c-5p/LGR4/beta-catenin axis activity. J Cell Mol Med. 2022;26:2218-29.
- [CrossRef] [PubMed] [Google Scholar]




