


Translate this page into:
Knockdown of Galectin-3 confers myocardial protection against ischemia-reperfusion injury, modulating oxidative stress, inflammatory response, and the peroxisome proliferator-activated receptor g signaling pathway

*Corresponding author: Xuehong Lin, Department of Cardiovascular Medicine, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, Shandong, China. linxueh2013@163.com
-
Received: ,
Accepted: ,
How to cite this article: Chen D, Wen J, Zang W, Lin X. Knockdown of Galectin-3 confers myocardial protection against ischemia-reperfusion injury, modulating oxidative stress, inflammatory response, and the peroxisome proliferator-activated receptor g signaling pathway. CytoJournal. 2025;22:49. doi: 10.25259/Cytojournal_12_2025
Abstract
Objective
Ischemia-reperfusion (I-R) injury in the myocardium is a considerable challenge in cardiovascular medicine, posing a severe threat to life. Given that galectin-3 possibly regulates myocardial I-R damage, this study aims to investigate the detailed mechanisms underlying galectin-3’s effects on myocardial I-R injury.
Material and Methods
The expression levels of galectin-3 in vivo and in vitro myocardial I-R models were determined by Western blot and quantitative real-time polymerase chain reaction. The effects of galectin-3 on inflammatory factors and oxidative stress factors in myocardial I-R were measured with an enzyme-linked immunosorbent assay, and the extent of myocardial tissue damage was assessed using hematoxylin-eosin staining. The influence of galectin-3 on peroxisome proliferator-activated receptor g (PPARg) signaling pathway-related proteins in myocardial I-R was determined by Western blot.
Results
Myocardial I-R damage was associated with increased galectin-3 expression, and the blood levels of creatine kinase-myocardial band and creatine kinase were favorably correlated with the messenger RNA levels of galectin-3 in mice with cardiac I-R damage. The inhibition of galectin-3 alleviated oxidative stress and inflammatory response, and galectin-3 promoted reactive oxygen species production in myocardial I-R cells. Furthermore, the cardiac I-R damage mouse model exhibited decreased expression of proteins linked to the PPARg signaling pathway, but galectin-3 inhibition enhanced the expression of these proteins.
Conclusion
Galectin-3 plays a crucial role in exacerbating myocardial I-R injury, and its up-regulation is associated with increased oxidative stress, inflammatory responses, and inhibition of the protective PPARg signaling pathway. The alleviation of these harmful effects by galectin-3 inhibition suggests that targeting galectin-3 is a potential therapeutic method for reducing myocardial I-R injury.
Keywords
Galectin-3
Inflammatory response
Myocardial ischemia-reperfusion injury
Oxidative stress
Peroxisome proliferator-activated receptor g

INTRODUCTION
Myocardial ischemia-reperfusion (I-R) injury is a critical event in cardiovascular diseases, greatly contributing to morbidity and mortality worldwide.[1,2] Although restoring blood flow to ischemic myocardial tissues is essential for salvaging viable heart muscles, this process often exacerbates tissue damage, which is collectively termed reperfusion injury.[3,4] Reperfusion injury encompasses complex biochemical and molecular processes, including oxidative stress, inflammatory reactions, and modifications to cellular signaling pathways. These factors lead to cardiomyocyte death and impaired cardiac function.[5,6]
Galectin-3 is a member of the b-galactoside-binding protein family and has attracted considerable interest.[7,8] Cell adhesion, apoptosis, and immunological responses are all affected by galectin-3, which is extensively expressed in a variety of cell types, including immune cells and cardiomyocytes.[9,10] Moreover, galectin-3 is considered a key mediator in heart failure and myocardial fibrosis, and its elevated levels are associated with adverse cardiac remodeling and poor clinical outcomes.[11,12]
Although galectin-3’s pathogenic significance in chronic cardiac diseases has been thoroughly investigated, its function in acute myocardial I-R damage is still unknown. Emerging evidence suggests that Galectin-3 may influence the extent of myocardial damage and functional recovery following I-R injury through the modulation of oxidative stress, inflammatory responses, and cellular signaling pathways. Specifically, galectin-3 is involved in the regulation of the peroxisome proliferator-activated receptor g (PPARg) signaling pathway, which plays a critical role in cellular metabolism, inflammation, and myocardial protection.[13]
This study aims to investigate the cardioprotective effects of galectin-3 knockdown in myocardial I-R injury and explore the mechanisms involving oxidative stress, inflammatory responses, and the PPARg signaling pathway. We hypothesize that galectin-3 knockdown can alleviate I-R-induced myocardial damage by reducing oxidative stress and inflammatory responses while enhancing PPARg signaling, thereby promoting cardiomyocyte survival and functional recovery. Insight into these processes can lead to the discovery of novel therapeutic targets for reducing myocardial I-R damage and enhancing the clinical results of ischemic heart disease patients.
MATERIAL AND METHODS
Myocardial I-R animal model
Male C57BL/6J mice (8-10 weeks, 20-25 g) were purchased from Aniphe Biolab (Nanjing, China) to establish the myocardial I-R injury model. The mice were kept at 23 ± 1 °C, subjected to 12 h light-12 h dark cycles, and received food and water ad libitum. Healthy male C57BL/6J mice were fasted for 12 h before surgery but were allowed to drink freely. After 2% pentobarbital sodium (50 mg/kg, 21642-83-1, Shandong Xiya Chemical Industry Co., Ltd., Shandong, China) was administered as an anesthetic, each mouse was secured on a surgical table, subjected to endotracheal intubation, and connected to a small-animal ventilator set at a respiratory rate of 90-120 breaths/min and tidal volume of 0.2-0.3 mL. After the disinfection of the left chest area, an incision was made through the skin and muscle layers between the fourth or fifth ribs on the left side to expose the ribs and heart.
Then, 6-0 sutures were used to ligate below the left anterior descending coronary artery, and the ligature was positioned 1-2 mm below the left atrial appendage. Successful ischemia was confirmed by observing the whitening of the anterior wall of the heart and considerable elevation of the ST segment on the electrocardiogram. Ischemia was maintained for 30-45 min, then coronary artery blood flow was restored by carefully releasing the ligature, and reperfusion was initiated. These procedures were performed for 1-2 h. After surgery, close the chest and skin layers with layered sutures to prevent pneumothorax. The mice recovered in a warm and quiet environment and were closely monitored for physiological parameters, including body temperature, respiratory rate, and heart rate. In addition to the 4 mice that died during the modeling process, a total of 24 mice were used for follow-up studies in this study, with six mice in each group.[14] Animals are grouped as follows:
Sham: This negative control group was composed of mice without any treatment.
I-R: The group was composed of mice with myocardial I-R.
-
I-R+Gal-3: Human recombinant galectin-3 (450-38-50UG, Peprotech, Rocky Hill, NJ) was administered with 5 mg diluted in an equal volume of saline.[15] Take an IV for 3 days.
I-R+anti-Gal-3: 5 mg/kg galectin-3 inhibitors G3-C12 (HY-P1592, MedChemExpress, Monmouth Junction, NJ, USA) was administered intravenously once a day for 3 days.[16]
After the experiment, all mice were anesthetized with 1% pentobarbital sodium and sacrificed for neck dislocation.
Cardiac ultrasound examination
Forty-eight hours after reperfusion, echocardiography was performed using an ultrasound machine, and 2% isoflurane (HY-A0134, MedChemExpress, Monmouth Country, NJ, USA) inhalation anesthesia was administered. Left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), left ventricular ejection fraction (LVEF), and left ventricular fractional shortening (LVFS) were obtained from M-mode echocardiograms captured in the muscle-level parasternal long-axis view and short-axis view.
Hematoxylin and eosin (HE) staining
First, the collected heart tissues were fixed in 4% paraformaldehyde (P0099, Beyotime Biotechnology, Shanghai, China) for 48 h. Then, gradually dehydrate the tissue using different concentrations of ethanol. Next, immerse the tissue in the clearing agent xylene to make it transparent, and then embed it in molten paraffin. After embedding, the tissues were cut into 5-10 mm-thick sections with a microtome, and the sections were sequentially immersed in HE staining solutions (BL700A, Biosharp Life Science, Hefei, Anhui, China) for 3 min. After staining, the sections were dehydrated, immersed in a clearing agent, mounted on slides with an organic solvent, and sealed with a mounting medium. Once staining is complete, place the slides under a microscope (BX46, Olympus Corporation, Tokyo, Japan) and observe the cell and tissue structures and the quality of staining at both low and high magnification.
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA integrity and purity were ensured by extracting the total RNA from the cell or tissue samples with TRIzol (R0016, Beyotime Biotechnology, Shanghai, China) and a spectrophotometer (Multiskan SkyHigh, Thermo Fisher Scientific, Waltham, MA, USA) was used in evaluating RNA concentration and purity. Reverse transcriptase (14604ES, Yeasen, Shanghai, China) was used to convert RNA into complementary DNA (cDNA). Then, a quantitative real-time polymerase chain reaction (qPCR) reaction mixture was prepared, which included the cDNA template, specific primers, qPCR fluorescent dye (such as SYBR Green) or probe, qPCR buffer, dNTPs, and Taq DNA polymerase (RR019B, TaKaRa Bio Inc., Kafu City, Yamanashi Prefecture, Japan). The reaction mixture was added to the qPCR reaction plate. After the reaction, the specificity of the amplified products was confirmed through melting curve analysis. Then, the fluorescence signals were analyzed using a qPCR instrument (QuantStudio, Thermo Fisher Scientific, Waltham, MA, USA). The cycle threshold (Ct) values were obtained. A low Ct value indicated the high initial copy number of the target gene. The 2−ΔΔCt method was used for relative quantification and calculation of change in the expression of a target gene relative to glyceraldehyde-3-phosphate dehydrogenase. The reliability of the results was confirmed through repeated experiments. The primer sequences used in this part of the experiment are shown in Table 1.
| Prime name | Prime sequences (5'-3') |
|---|---|
| Human | |
| Galectin-3-F | CAAAGAGGGAATGATGTTGCC |
| Galectin-3-R | TGTTCTCATTGAAGCGTGGGT |
| IFN-γ-F | CAGCTCTGCATCGTTTTGGG |
| IFN-γ-R | GTTCCATTATCCGCTACATCTGAA |
| TNF-α-F | AGGCGCTCCCCAAGAAGACA |
| TNF-α-R | AAGTGCAGCAGGCAGAAGAG |
| GAPDH-F | CCATGTTCGTCATGGGTGTG |
| GAPDH-R | GGTGCTAAGCAGTTGGTGGTG |
| Mouse | |
| Galectin-3-F | CACCTGCACCTGGAGTCTAC |
| Galectin-3-R | GCACTGGTG AGGTCTATGTC |
| IFN-γ-F | GATGGTGACATGAAAATC |
| IFN-γ-R | GGCAATACTCACGAATGC |
| TNF-α-F | GAGCACAGAAAGCATGATCCG |
| TNF-α-R | CTCCTCCACTTGGTGGTT TGC |
| GAPDH-F | TGAAGGTCGGTGTGAACGGATTTGGC |
| GAPDH-R | CATGTAGGCCATGAGGTCCACCAC |
IFN-g: Interferon gamma, TNF-a: Tumor necrosis factor-alpha, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, A: Adenine, C: Cytosine, G: Guanine, T: Thymine, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
Cell culture
AC16 cardiomyocytes (iCell-h323) were purchased from iCell (Shanghai, China) and cultured in high-glucose Dulbecco’s modified eagle medium (iCell-0001, iCell, Shanghai, China) containing 10% fetal bovine serum (BL205A, Biosharp Life Science, Hefei, Anhui, China) and 1% penicillin-streptomycin (60162ES76, Yeasen, Shanghai, China). The culture environment was maintained at 37°C with 5% carbon dioxide (CO2). When the cells reached 70-80% confluency, they were passaged by removing the culture medium and washing with phosphate-buffered saline (PBS). Trypsin-ethylene diamine tetraacetic acid (EDTA, E8008, Gibco, Life Technologies, Rockville, MD, USA) solution was used, and once the cells detached, trypsin was neutralized with the culture medium. The cells were collected through centrifugation and resuspended. The cells were split in ratios of 1:3-1:6 into new culture flasks. For cryopreservation, cells in the logarithmic growth phase were collected through centrifugation, resuspended in a freezing medium, aliquoted into cryovials, and stored at −80°C overnight. The oxygen concentration in the cell culture medium was reduced to simulate ischemic conditions (1% oxygen, balanced CO2). Place the cells in a hypoxia chamber to ensure exposure to low oxygen conditions for 2 h. At the end of the hypoxic period, the cell culture medium was replaced with prewarmed ischemic buffer or oxygen-containing medium, and the cells were stored in an incubator at 37°C with 5% CO2 for reperfusion.[17] AC16 cardiomyocytes were mycoplasma-free, and short-tandem repeat analysis revealed that they were derived from parental cells. The cells were treated with GW9662:10mM for 30 min.[18]
Cell transfection
Overexpression plasmids containing galectin-3 gene (pCMVgalectin-3; sense, 5'-GGCTAGAAGCTTATGGCAGACAATTT TTCGCTG-3'; antisense, 5'-GGCGAATT CTTATATATGG TATATGA-3'), control plasmid (pCMV-NC), small interfering (siRNA), and siRNA-targeting galectin-3 (sense, 5'-GUACA AUCAUCGGGUUAAATT-3' and antisense, 5'-UUUAACCCGAUGAUUGUACTT-3') were purchased from Sangon (Shanghai, China). AC16 cells were plated into sterile culture dishes or flasks and cultured until they reached 70-80% confluence. Before transfection, the plasmid DNA was mixed with the transfection reagent (Lipofectamine 2000; 11668027, Invitrogen, Waltham, Massachusetts, USA) and incubated in a serum-free medium for 30 min. The mixture was dripped onto cells in a dish, gently rotated to distribute evenly, and incubated with the transfection reagent for 6 h.
Enzyme-linked immunosorbent assay
Assay kits for creatine kinase (CK, A032-1-1), creatine kinase-myocardial band (CK-MB, H197-1-1), interleukin (IL)-1b (H002-1-2), IL-6 (H007-1-1), reactive oxygen species (ROS, E004-1-1), superoxide dismutase (SOD, A001-3-2), malondialdehyde (MDA, A003-4-1), glutathione (GSH, A006-2-1), and glutathione peroxidase (GSH-Px, A005-1-2) were obtained from Jiancheng Bioengineering Institute (Nanjing, China). Cell culture supernatants and myocardial tissues were collected, and tissue homogenates or cell lysates were prepared. The absorbance of samples was measured at specific wavelengths with an enzyme-linked immunosorbent assay reader (ELx405, BioTeK, Winooski, Vermont, USA).
Western blot
Proteins were extracted from cell or tissue samples with a lysis buffer (R0010, Solarbio, Beijing, China). Protein quantification methods were used in measuring the extracted protein concentration, ensuring equal loading of protein into the gel. Protein samples were mixed with a loading buffer (P1040, Solarbio, Beijing, China), heated for protein denaturation, and loaded onto polyacrylamide gels. Electrophoresis was conducted to separate proteins by size and charge into bands. The separated proteins were then transferred through electrophoresis onto polyvinylidene difluoride membranes (IPVH00010, Millipore Corporation, Billerica, MA, USA). The membranes were blocked with 5% bovine serum albumin (I017-1-1, Jiancheng Bioengineering Institute, Nanjing, China) for 1 h, and the primary antibody (1:1000, CST, BSN, Ma, USA) was incubated overnight at 4°C. Then, the membrane was washed 3 times with tris buffered saline with tween-20 (TBST), and the secondary antibody (1:10000, #14708, CST, BSN, Ma, USA) was incubated for 1 h at room temperature. The presence of specific protein bands was detected by chemiluminescence staining (BL520b, Biosharp Life Science, Hefei, Anhui, China). Gel imaging systems (ChemiDoc XRS+ System, Bio-Rad Laboratories, Hercules, California, USA) were employed to capture images. ImageJ software (Version 1.5e, National Institutes of Health, Bethesda, Maryland) was used in analyzing the imaging results, band intensities were measured and compared, and the expression levels of the target protein were evaluated.
The primary antibodies were galectin-3 (#89572), PPAPg (#2443), heme oxygenase-1 (HO-1, #43966), nuclear factor erythroid 2-related factor 2 (Nrf2, #20733), glutathione peroxidase 4 (GPx4, #59735), and GAPDH (#2118).
ROS assay
ROS production in cultured cells was quantified by cell permeability fluorescence probe 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA). Then, 1 × 105 cells were stained with 10 mM DCFH-DA (50101ES01, Yeasen, Shanghai, China) at 37°C for 30 min, then separated with trypsin/EDTA (25200056, Thermo Fisher Scientific, Waltham, MA, USA), washed, and resuspended in PBS for flow cytometry (FACS Aria III, BD bioscience, Franklinhoo, New Jersey, USA).
Statistical analysis
The experimental data underwent analysis through GraphPad Prism 9.5 software (GraphPad Software Inc, California, USA), and the findings were presented as mean ± standard deviation. Discrepancies were scrutinized using unpaired two-sided Student’s t-test for two-group comparisons or a one-way analysis of variance test followed by a Turkey post hoc test for multiple comparisons. Statistical significance was established at P < 0.05.
RESULTS
Galectin-3 is upregulated in myocardial I-R injury
Figure 1 shows the expression pattern of galectin-3 in a myocardial I-R model. Compared with the sham surgery group, the group with induced myocardial I-R showed upregulated messenger RNA (mRNA) and protein levels of galectin-3 in myocardial tissues (P < 0.001); [Figure 1a-c].
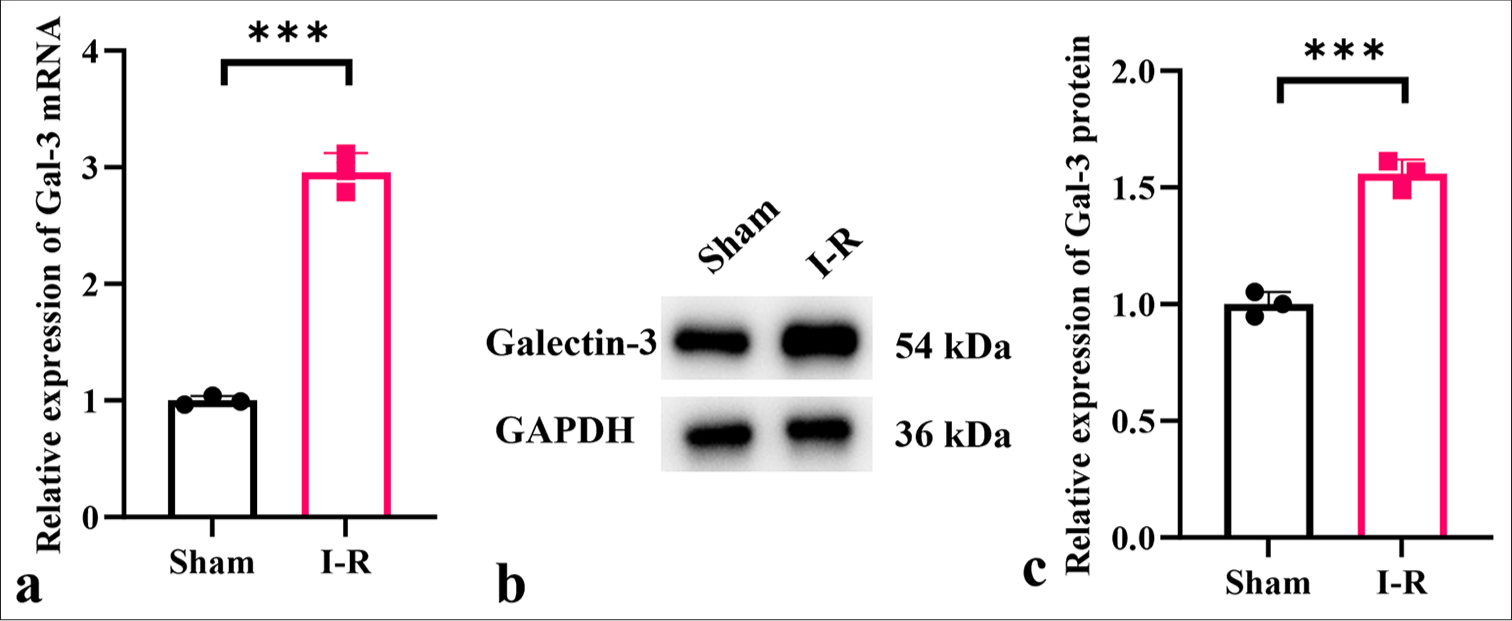
- Expression levels of galectin-3 in myocardial I-R injury. (a-c) mRNA and protein expression levels of galectin-3 in myocardial I-R injury mouse tissues. (n = 3, ✶✶✶P < 0.001). I-R: Ischemiareperfusion, Gal: galectin, I-R: Ischemia-reperfusion, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, mRNA: Messenger RNA.
Galectin-3 enhances oxidative stress and inflammatory response in an in vivo myocardial I-R model
To elucidate the role of galectin-3 in the in vivo myocardial I-R model, we injected recombinant human galectin-3 protein into myocardial I-R mice. Human galectin-3 recombinant protein increased the CK-MB and CK levels of the myocardial I-R mice, exacerbating myocardial injury, elevating LVEDD, LVESD, and LVFS levels, and reducing LVEF levels (P < 0.001) [Figure 2a-f]. Human galectin-3 recombinant protein elevated the levels of interleukin 1 beta (IL-1b), interleukin 6 (IL-6), interferon gamma (IFN-g), and tumor necrosis factor alpha (TNF-a) and increased the levels of MDA in mouse heart tissues while reducing SOD, GSH, and GSH-px activity (P < 0.001) [Figure 2g-n]. HE staining results showed that I-R+Gal-3 considerably exacerbated myocardial tissue damage and increased cellular injury [Figure 2o].
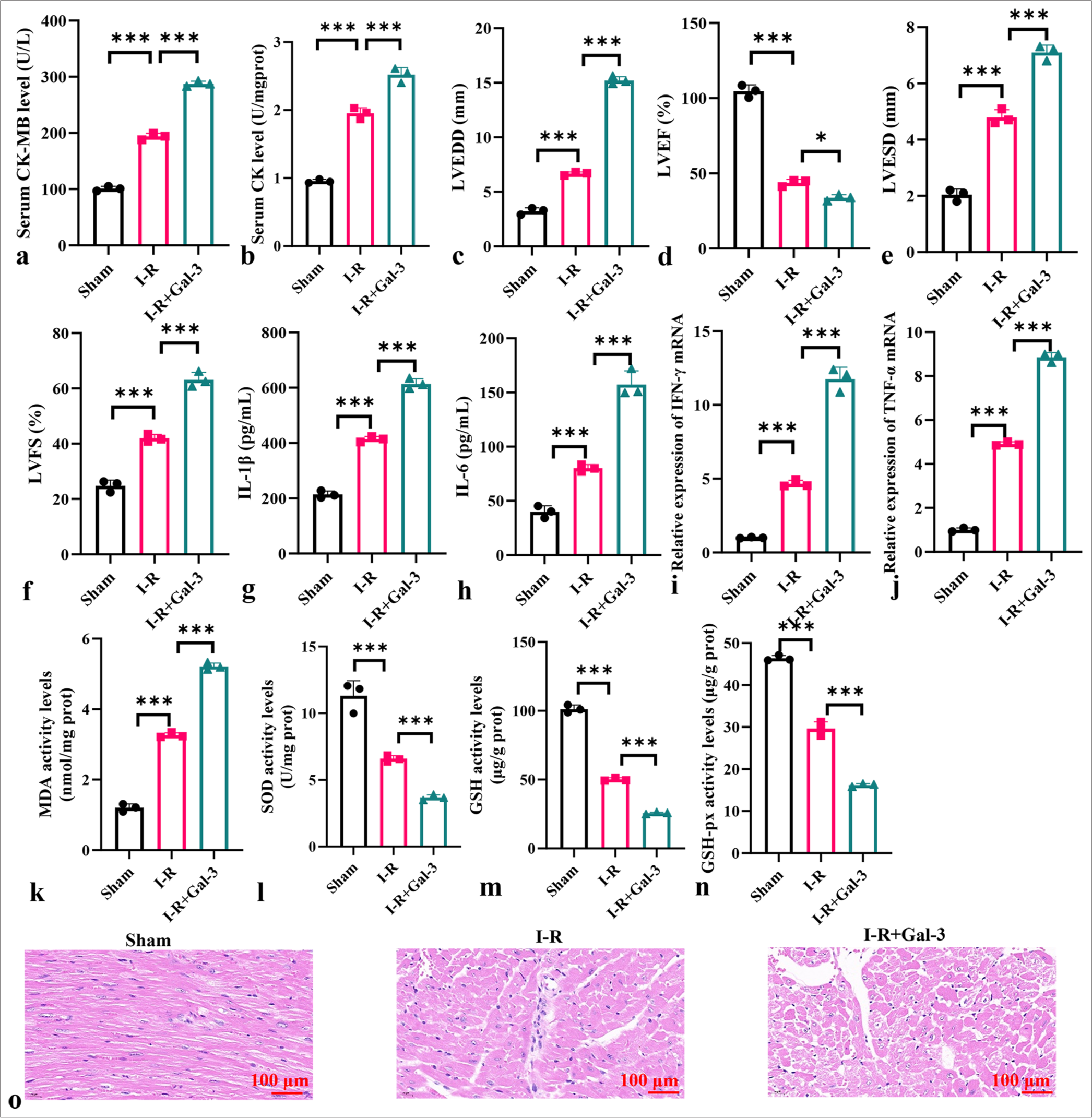
- Galectin-3 enhances oxidative stress and inflammatory response in an in vivo myocardial I-R model. (a and b) Levels of CK-MB (a) and CK (b) in serum. (c) LVEDD. (d) LVEF. (e) LVESD. (f) LVFS. (g-j) Levels of IL-1b, IL-6, TNF-a, and IFN-g in myocardial I-R tissues. (k-n) Levels of MDA, SOD, GSH, and GSH-px in myocardial tissues. (o) HE staining of myocardial I-R tissues. Objective: ×200. Scale bar = 100 μm. (n = 3, ✶P < 0.05, ✶✶✶P < 0.001). I-R: Ischemia-reperfusion, CK: Creatine kinase, CK-MB: creatine kinasemyocardial band, LVEF: Left ventricular ejection fraction, LVESD: Left ventricular end-systolic dimension, LVFS: Left ventricular fractional shortening, LVEDD: Left ventricular end-diastolic diameter, IL: Interleukin, TNF-a: Tumor necrosis factor alpha, IFN-g: Interferon gamma, MDA: Malondialdehyde, SOD: Superoxide dismutase, GSH: Glutathione, GSH-Px: Glutathione peroxidase, HE: Hematoxylin-eosin.
Anti-galectin-3 inhibits oxidative stress and inflammatory response in myocardial I-R model
The anti-galectin-3 body reduced the levels of CK-MB and CK in the sera of myocardial I-R mice, alleviated myocardial injury, decreased LVEDD, LVESD, and LVFS levels, and increased LVEF levels (P < 0.001) [Figure 3a-f]. Anti-galectin-3 body lowered the levels of IL-1b, IL-6, IFN-g, and TNF-a in the mouse myocardial tissues and reduced MDA levels while increasing SOD, GSH, and GSH-px activity (P < 0.001) [Figure 3g-n]. HE staining results showed that I-R+anti-Gal-3 considerably reduced myocardial tissue damage and decreased cellular injury [Figure 3o].
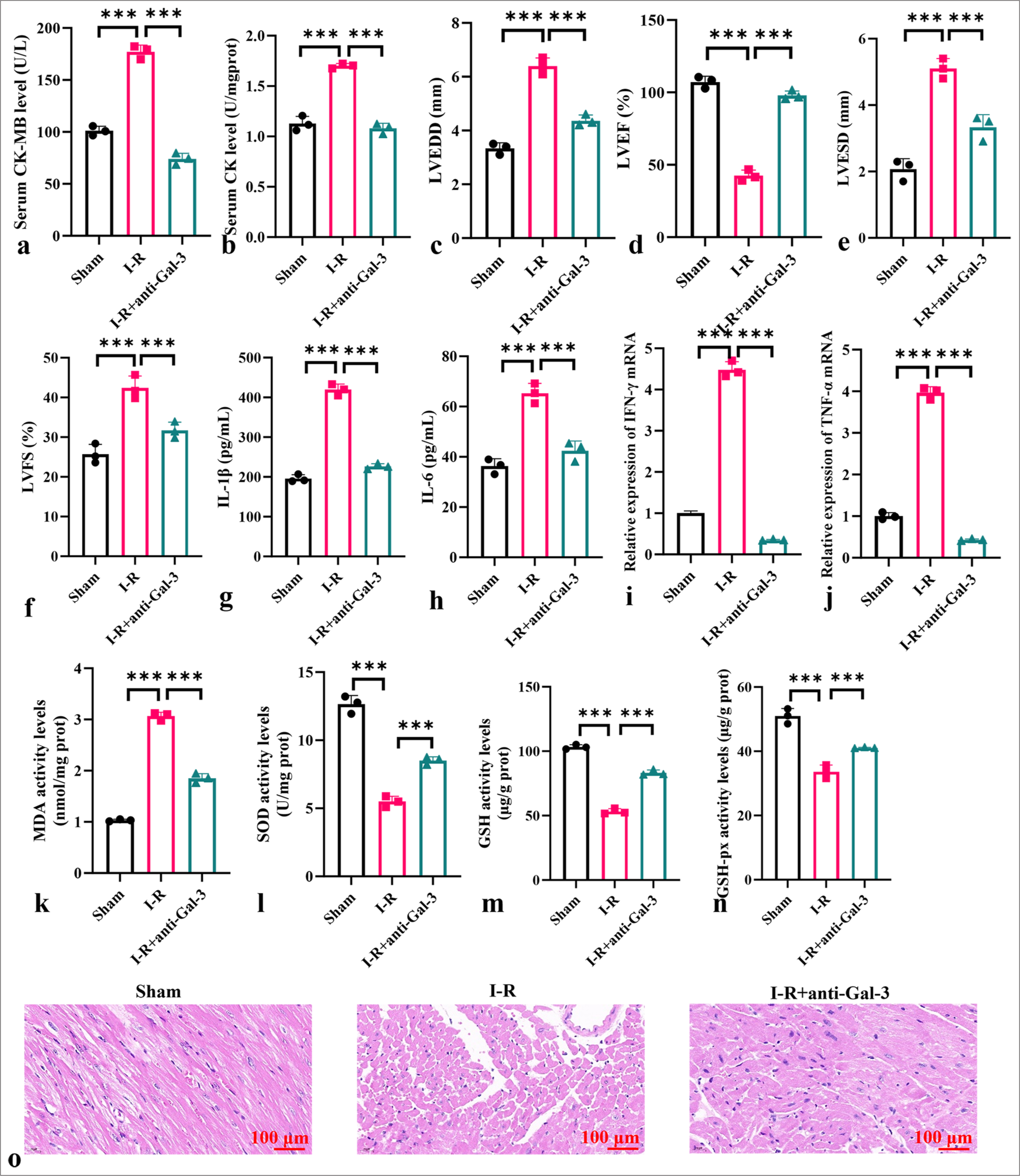
- Anti-galectin-3 attenuates oxidative stress and inflammatory response in myocardial I-R model. (a and b) Levels of CK-MB (a) and CK (b) in serum. (c) LVEDD. (d) LVEF. (e) LVESD. (f) LVFS. (g-j) Levels of IL-1b, IL-6, TNF-a, and IFN-g in myocardial tissues. (k-n) Levels of MDA, SOD, GSH, and GSH-px in myocardial tissues. (o) HE staining of myocardial I-R tissues. Objective: ×200. Scale bar = 100 μm. (n = 3, ✶✶✶P < 0.001). I-R: Ischemia-reperfusion, CK: Creatine kinase, CK-MB: creatine kinase-myocardial band, LVEF: Left ventricular ejection fraction, LVESD: Left ventricular end-systolic dimension, LVFS: Left ventricular fractional shortening, LVEDD: Left ventricular enddiastolic diameter, IL: Interleukin, TNF-a: Tumor necrosis factor alpha, IFN-g: Interferon gamma, MDA: Malondialdehyde, SOD: Superoxide dismutase, GSH: Glutathione, GSH-Px: Glutathione peroxidase, HE: Hematoxylin-eosin.
Experimental study on galectin-3 regulation of myocardial I-R-induced in vitro inflammatory response
Galectin-3 expression plasmid or si-galectin-3 mimic was used to modulate galectin-3 expression in an in vitro myocardial I-R model. The silencing or overexpression of galectin-3 was successful (P < 0.001) [Figure 4a-d]. Furthermore, in the in vitro myocardial I-R model, galectin-3 overexpression increased the levels of IL-1b, IL-6, IFN-g, and TNF-a (P < 0.001) [Figure 4e-h]. The down-regulation of galectin-3 inhibited the levels of these cytokines (P < 0.001) [Figure 4i-l]. In Figure 4m-p, the overexpression of galectin-3 downregulated the expression of PPARg, HO-1, Nfr2, and GPX4. By contrast, the expression of these proteins was up-regulated after galectin-3 silencing and decreased after treatment with the PPARg inhibitor GW9662.
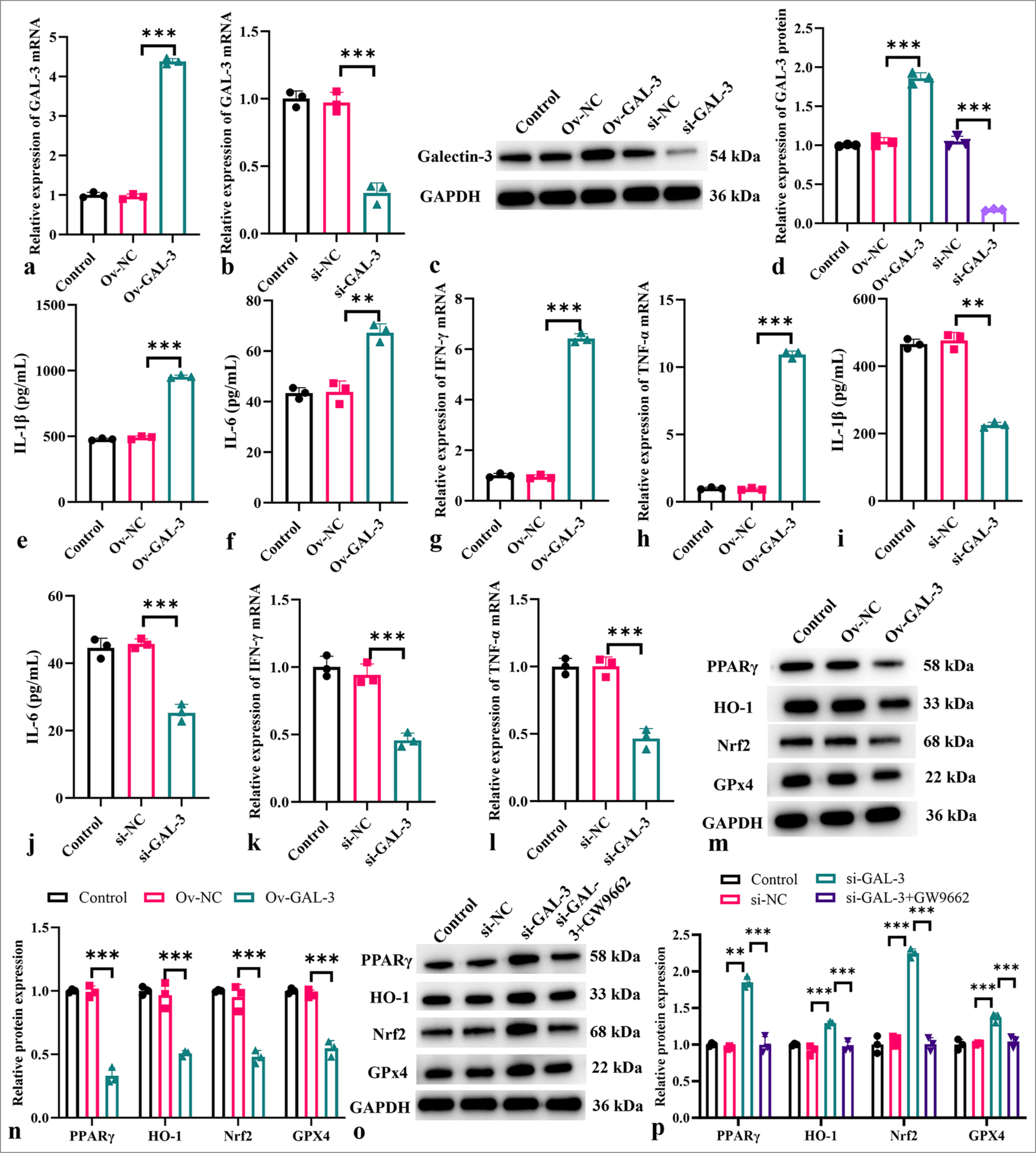
- Modulatory role of galectin-3 on inflammation in an in vitro myocardial I-R model. (a) Measurement of galectin-3 mRNA levels after transfection with the galectin-3 overexpression plasmid. (b) Measurement of galectin-3 mRNA levels after si-galectin-3 transfection. (c and d) Measurement of galectin-3 protein levels after transfection with the galectin-3 overexpression plasmid or si-galectin-3 transfection. (e-h) Levels of IL-1b (e), IL-6 (f), IFN-g (g), and TNF-a (h) after galectin-3 overexpression. (i-l) Levels of IL-1b (i), IL-6 (j), IFN-g (k), and TNF-a (l) after galectin-3 knockdown. (m-p) Protein expression levels of PPARg, HO-1, Nrf2, and GPx4 measured in different treatment groups. (n = 3, ✶✶P < 0.01, ✶✶✶P < 0.001). I-R: Ischemia-reperfusion, mRNA: Messenger RNA, IL: Interleukin, TNF-a: Tumor necrosis factor alpha, IFN-g: Interferon gamma, PPARg: Peroxisome proliferator-activated receptor g, HO-1: Heme oxygenase-1, Nrf2; Nuclear factor erythroid 2-related factor 2, GPx4: Glutathione peroxidase 4.
Galectin-3 affects ROS levels in myocardial I-R cells
In Figures 5a and b, galectin-3 silencing considerably inhibited ROS production in the I-R cells (P < 0.001). In Figures 5c and d, the ROS levels increased considerably after the overexpression of galectin-3 (P < 0.001). According to these findings, galectin-3 and ROS production in the myocardial I-R model may be tightly connected.
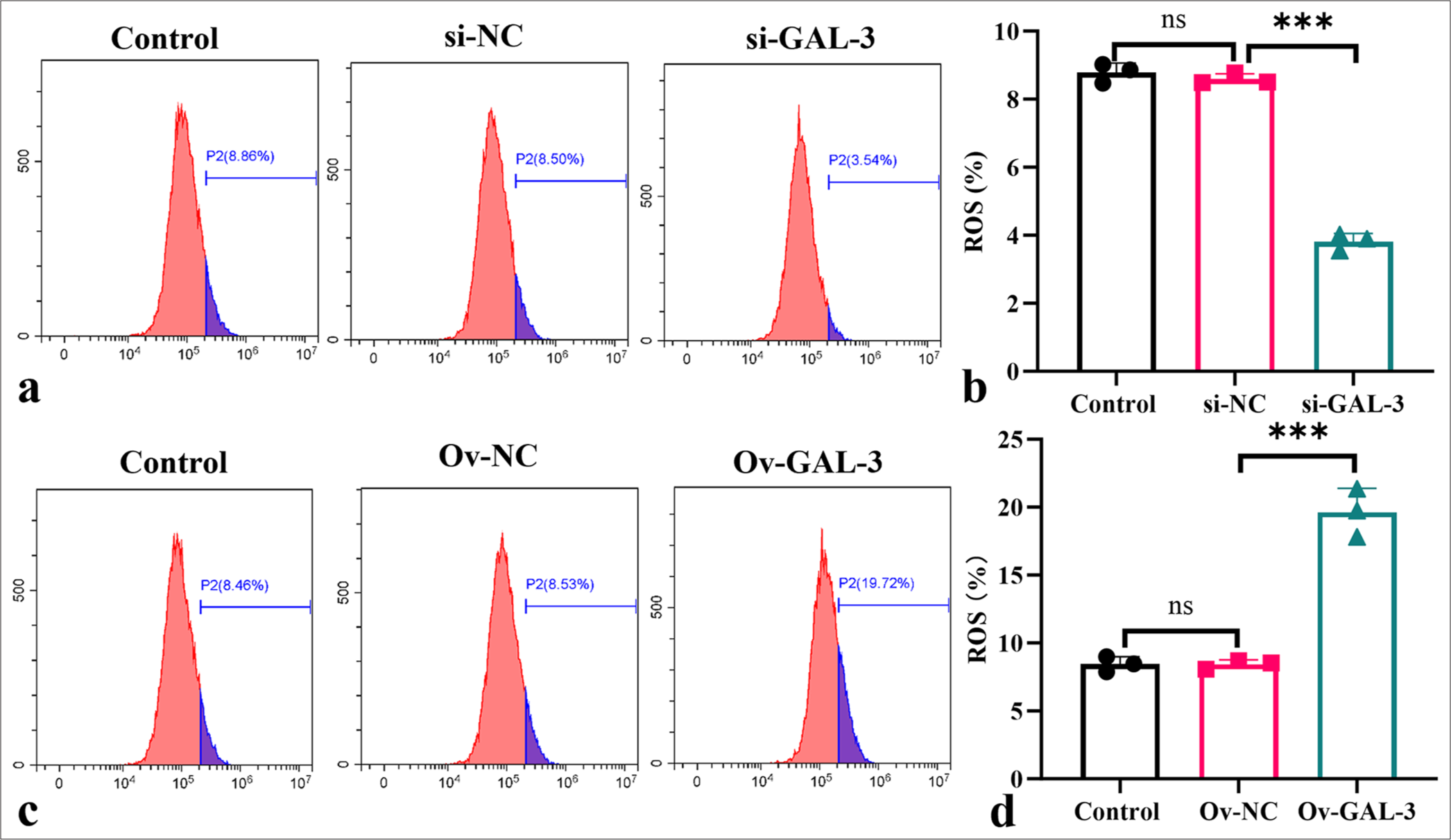
- Galectin-3 promotes ROS production in the myocardial I-R cells. (a-d) Flow cytometry was used in determining ROS levels in the myocardial I-R cells after silencing (a and b) or overexpressing galectin-3 (c and d). (n = 3, ns: no significant difference, ✶✶✶P < 0.001). I-R: Ischemia-reperfusion.
Galectin-3 regulates the PPARg signaling pathway in myocardial I-R injury
Figures 6a-e indicate that compared with the sham group, the I-R group protein showed considerably decreased PPARg, HO-1, Nrf2, and GPx4 levels (P < 0.001). The I-R+Gal-3 group showed lower levels of these proteins (P < 0.001), while the I-R+anti-Gal-3 group exhibited significantly elevated protein levels of these proteins (P < 0.001).
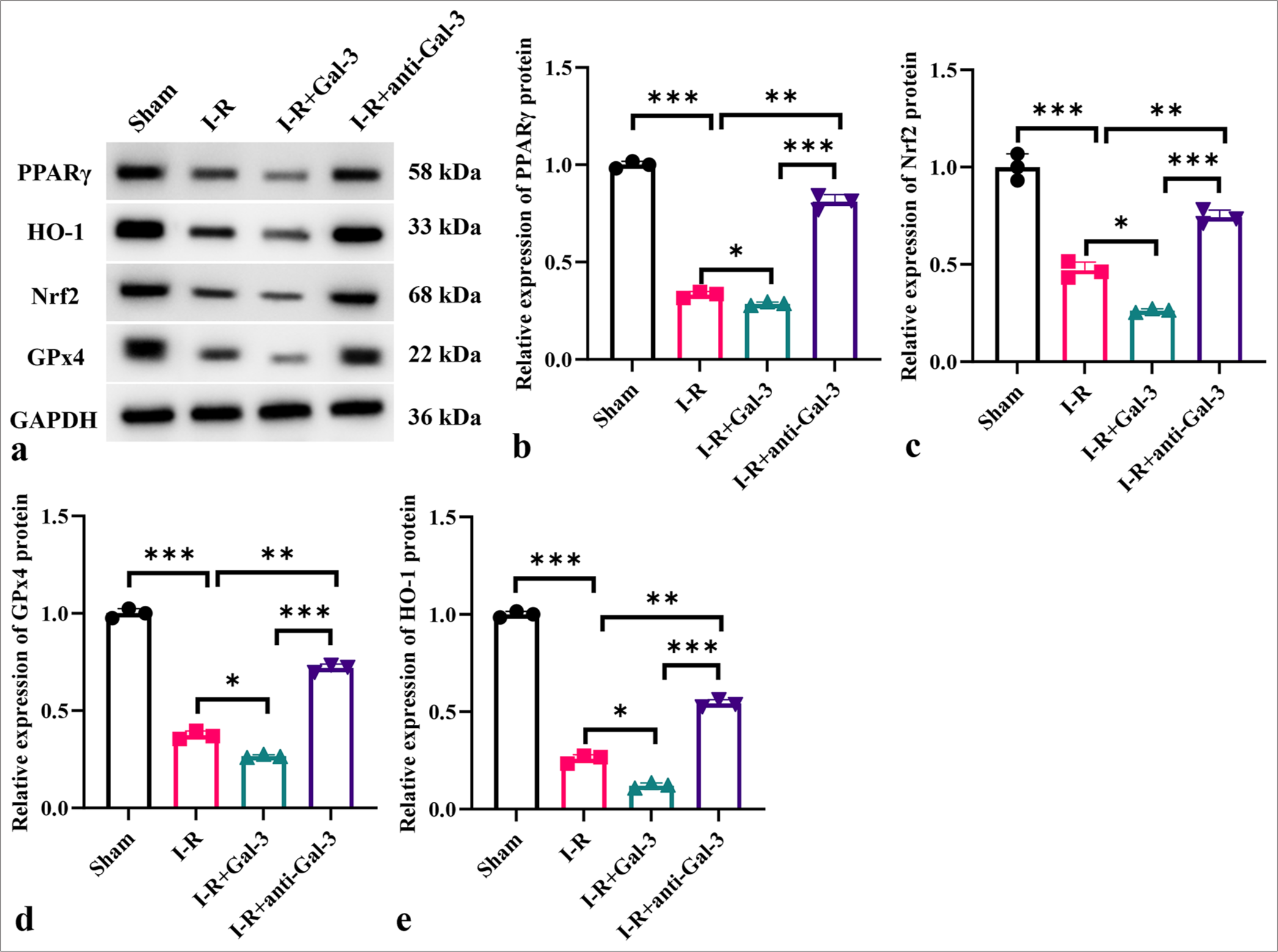
- Galectin-3 regulates the PPARg signaling pathway in myocardial I-R injury. (a-e) Protein expression levels of PPARg, HO-1, Nrf2, and GPx4 in different treatment groups. (n = 3, ✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001). I-R: Ischemia-reperfusion, PPARg: Peroxisome proliferator-activated receptor g, HO-1: Heme oxygenase-1, Nrf2; Nuclear factor erythroid 2-related factor 2, GPx4: Glutathione peroxidase 4.
DISCUSSION
This study demonstrated that myocardial I-R injury greatly increased the expression of galectin-3 and that adjusting galectin-3 levels influenced the degree of cardiac damage and inflammatory reactions. The findings demonstrated that by encouraging oxidative stress and inflammation in the I/R model, galactin-3 blocked protective signaling pathways like PPARg.
Animals with I-R damage showed considerably increased galectin-3 mRNA and protein levels in their cardiac tissues. Increased blood levels of myocardial damage indicators CK-MB and CK, increased levels of LVEDD and LVESD, and lowered levels of LVEF were linked to elevated galectin-3 levels. These results demonstrated that, in the context of I-R injury, galectin-3 is both a marker and a mediator of myocardial damage.
The injection of recombinant human galectin-3 protein into the myocardial I-R model further exacerbated myocardial injury. Pro-inflammatory cytokine levels were up-regulated, MDA levels were elevated, and the antioxidant enzymes SOD, GSH, and GSH-px were less active in the cardiac tissues. The HE staining results supported these findings, showing severe cellular damage in the I-R+Gal-3 group.
Meanwhile, the degree of myocardial damage was reduced in the I-R model after the administration of an anti-galectin-3 antibody. The treatment reduced the serum levels of CKMB and CK, decreased LVEDD, LVESD, and LVSD levels, and increased LVEF levels. It also lowered the levels of pro-inflammatory cytokines and MDA while increasing the activity of antioxidant enzymes. Reduced cellular damage in the I-R+anti-Gal-3 group was verified by HE staining, indicating that blocking galectin-3 is a protective strategy against myocardial I-R injury.
In vitro experiments further confirmed the role of galectin-3 in modulating inflammatory responses. The overexpression of galectin-3 in the myocardial I-R model increased the levels of pro-inflammatory cytokines and reduced cell infiltration, and galectin-3 down-regulation decreased their levels. In addition, after galectin-3 overexpression, muscle fiber disturbance and coagulation necrosis were reduced, and interstitial edema and large area necrosis were reduced. These results highlighted the pro-inflammatory role of galectin-3 in myocardial I-R injury.
The PPARg signaling pathway was found to be significantly affected by galectin-3 modulation. The protein levels of PPARg, HO-1, Nrf2, and GPx4 were markedly reduced in the I-R group. Moreover, galectin-3 overexpression further decreased the levels of these protective proteins, whereas anti-galectin-3 treatment increased their levels. Nrf2 transcription factor regulates oxidative stress response to maintain the dynamic balance of intracellular REDOX. Due to KEAP1-mediated proteasome degradation, Nrf2 is a low-abundance protein under non-stress conditions.[19] Our results showed that galectin-3 enhanced Nrf2 translocation and increased the expression of its downstream effector HO-1, consistent with previous research.[20] The results suggested that the PPARg signaling pathway, which has anti-inflammatory and antioxidant properties, is negatively regulated by galectin-3.
Our findings align with previous studies that have demonstrated the deleterious role of galectin-3 in cardiac injury and remodeling.[21,22] For instance, elevated galectin-3 levels are associated with adverse outcomes in heart failure and myocardial fibrosis. This finding supports the notion that galectin-3 contributes to cardiac pathology through its pro-inflammatory and pro-fibrotic actions.[23,24]
Galectin-3’s role in increasing oxidative stress and inflammation in the context of myocardial I-R damage has been explored.[25,26] Galectin-3 deficiency in mice led to reduced infarct size and improved cardiac function after I-R injury. This finding is consistent with our findings, which demonstrated that inhibiting galectin-3 attenuates myocardial damage and enhances cardiac function.[27]
Our study enhances our understanding of the mechanisms by which galectin-3 modulates cardiac injury by demonstrating its impact on the PPARg signaling pathway. PPARg’s anti-inflammatory and antioxidant properties provide protection against I-R damage.[28-30] Our findings showed that galectin-3 negatively regulates PPARg and its downstream targets HO-1, Nrf2, and GPx4, providing novel insights into how galectin-3 exacerbates myocardial injury and suggesting that galectin-3 impairs the protective effects of PPARg signaling and thereby promotes oxidative stress and inflammation.
However, this study has some limitations. First, this study did not use clinical samples for verification, and thus, determining the safety and effectiveness of the current conclusions in clinical practice is impossible. Second, this study did not further explore how galectin-3 specifically affects the PPARg pathway, nor did it explore other possible pathways. In subsequent studies, the clinical samples of myocardial ischemia will be used, and the in-depth associations among galectin-3, PPARg, and other pathways of galectin in cardiovascular disease will be explored.
In summary, our findings are consistent with and extend the existing body of literature on the role of galectin-3 in myocardial injury. Targeting galectin-3 with specific antibodies or down-regulating its expression is a potential therapeutic strategy for mitigating myocardial I-R injury and improving cardiac function. To determine the exact mechanisms by which galectin-3 controls these pathways and to assess the potential of galectin-3 inhibitors in therapeutic settings, more research is required.
SUMMARY
This work demonstrates that galectin-3 plays a crucial role in exacerbating myocardial I-R injury by increasing oxidative stress and inflammation and inhibiting protective signaling pathways like PPARg.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the present study were available from the corresponding author on reasonable request.
ABBREVIATIONS
I-R: Ischemia-reperfusion
PPARγ: Peroxisome proliferator-activated receptor γ
LVEDD: Left ventricular end-diastolic diameter
LVESD: Left ventricular end-systolic diameter
LVEF: Left ventricular ejection fraction
LVFS: Left ventricular fractional shortening
HE: Hematoxylin and eosin
qRT-PCR: Quantitative real-time polymerase chain reaction
IFN-γ: Interferon gamma
TNF-α: Tumor necrosis factor-alpha
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
PBS: Phosphate-buffered saline
EDTA: Ethylene diamine tetra acetic acid
CK: Creatine kinase
IL: Interleukin
ROS: Reactive oxygen species
SOD: Superoxide dismutase
MDA: Malondialdehyde
GSH: Glutathione
GSH-Px: Glutathione peroxidase
TBST: Tris buffered saline with tween-20
DCFH-DA: 2',7'-dichlorodihydrofluorescein diacetate
AUTHOR CONTRIBUTIONS
DC and WZ: Designed the study; all authors conducted the study; JY.W and XH.L: Collected and analyzed the data; XH.L and DC: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, took public responsibility for appropriate portions of the content, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work were appropriately investigated and resolved.
ACKNOWLEDGMENT
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Beijing Medconor Biotechnology Co., LTD. The study was approved by the Institutional Animal Care and Use Committee of Beijing Medconor Biotechnology Co., LTD(No. MDKN-2024-057). Informed consent to participate is not required, as this study does not involve human subjects.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: Not applicable.
References
- Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc Med. 2023;33:357-66.
- [CrossRef] [PubMed] [Google Scholar]
- Myocardial ischemiareperfusion injury; Molecular mechanisms and prevention. Microvasc Res. 2023;149:104565.
- [CrossRef] [PubMed] [Google Scholar]
- Nanomedicine-based therapeutics for myocardial ischemic/reperfusion injury. Adv Healthc Mater. 2023;12:e2300161.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammation in myocardial ischemia/reperfusion injury: Underlying mechanisms and therapeutic potential. Antioxidants (Basel). 2023;12:1944.
- [CrossRef] [PubMed] [Google Scholar]
- Ischemiareperfusion injury: Molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. 2024;9:12.
- [CrossRef] [PubMed] [Google Scholar]
- Ischemia reperfusion injury induced blood brain barrier dysfunction and the involved molecular mechanism. Neurochem Res. 2023;48:2320-34.
- [CrossRef] [PubMed] [Google Scholar]
- Novel galectin-3 roles in neurogenesis, inflammation and neurological diseases. Cells. 2021;10:3047.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting galectin-3 in inflammatory and fibrotic diseases. Trends Pharmacol Sci. 2023;44:519-31.
- [CrossRef] [PubMed] [Google Scholar]
- Galectin-3 aggravates pulmonary arterial hypertension via immunomodulation in congenital heart disease. Life Sci. 2019;232:116546.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical bone stem cells modify cardiac inflammation after myocardial infarction by inducing a novel macrophage phenotype. Am J Physiol Heart Circ Physiol. 2021;321:H684-701.
- [CrossRef] [PubMed] [Google Scholar]
- Modified citrus pectin ameliorates myocardial fibrosis and inflammation via suppressing galectin-3 and TLR4/MyD88/NF-kB signaling pathway. Biomed Pharmacother. 2020;126:110071.
- [CrossRef] [PubMed] [Google Scholar]
- Galectin-3, acute kidney injury and myocardial damage in patients with acute heart failure. J Card Fail. 2023;29:269-77.
- [CrossRef] [PubMed] [Google Scholar]
- Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020;7:588-603.
- [CrossRef] [PubMed] [Google Scholar]
- Resveratrol, novel application by preconditioning to attenuate myocardial ischemia/reperfusion injury in mice through regulate AMPK pathway and autophagy level. J Cell Mol Med. 2022;26:4216-29.
- [CrossRef] [PubMed] [Google Scholar]
- The role of galectin-3 and galectin-3-binding protein in venous thrombosis. Blood. 2015;125:1813-21.
- [CrossRef] [PubMed] [Google Scholar]
- Cardioprotective effects of galectin-3 inhibition against ischemia/reperfusion injury. Eur J Pharmacol. 2019;863:172701.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic maturation increases susceptibility to hypoxia-induced damage in human iPSC-derived cardiomyocytes. Stem Cells Transl Med. 2022;11:1040-51.
- [CrossRef] [PubMed] [Google Scholar]
- Akebia saponin D acts via the PPAR-gamma pathway to reprogramme a pro-neurogenic microglia that can restore hippocampal neurogenesis in mice exposed to chronic mild stress. CNS Neurosci Ther. 2023;29:2555-71.
- [CrossRef] [PubMed] [Google Scholar]
- Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89-116.
- [CrossRef] [PubMed] [Google Scholar]
- Kinsenoside mitigates myocardial ischemia/reperfusion-induced ferroptosis via activation of the Akt/Nrf2/HO-1 pathway. Eur J Pharmacol. 2023;956:175985.
- [CrossRef] [PubMed] [Google Scholar]
- The Role of biomarkers in cardiooncology. J Cardiovasc Transl Res. 2020;13:431-50.
- [CrossRef] [PubMed] [Google Scholar]
- Role of galectin-3 in cardiac dysfunction induced by subarachnoid hemorrhage. Exp Neurol. 2023;365:114418.
- [CrossRef] [PubMed] [Google Scholar]
- Biomarkers for the diagnosis and management of heart failure. Heart Fail Rev. 2022;27:625-43.
- [CrossRef] [PubMed] [Google Scholar]
- Does Galectin-3 (myocardial fibrosis biomarker) predict progression in chagas disease? Arq Bras Cardiol. 2021;116:257-58.
- [Google Scholar]
- Pentoxifylline treatment alleviates kidney ischemia/reperfusion injury: Novel involvement of galectin-3 and ASK-1/JNK & ERK1/2/NF-kB/HMGB-1 trajectories. J Pharmacol Sci. 2021;146:136-48.
- [CrossRef] [PubMed] [Google Scholar]
- Evaluating the protective role of trimetazidine versus nano-trimetazidine in amelioration of bilateral renal ischemia/reperfusion induced neuro-degeneration: Implications of ERK1/2, JNK and Galectin-3/NF-kB/TNF-a/HMGB-1 signaling. Tissue Cell. 2023;85:102241.
- [CrossRef] [PubMed] [Google Scholar]
- Recombinant human growth hormone treatment of mice suppresses inflammation and apoptosis caused by skin flap ischemiareperfusion injury. J Cell Biochem. 2019;120:18162-71.
- [CrossRef] [PubMed] [Google Scholar]
- Pharmacological activation of PPARb/q preserves mitochondrial respiratory function in ischemia/reperfusion via stimulation of fatty acid oxidation-linked respiration and PGC-1a/NRF-1 signaling. Front Endocrinol (Lausanne). 2022;13:941822.
- [CrossRef] [PubMed] [Google Scholar]
- Reno-and hepato-protective effect of allopurinol after renal ischemia/reperfusion injury: Crosstalk between xanthine oxidase and peroxisome proliferator-activated receptor gamma signaling. Food Chem Toxicol. 2023;178:113868.
- [CrossRef] [PubMed] [Google Scholar]
- Ischemia-reperfusion damage is attenuated by GQ-11, a peroxisome proliferator-activated receptor (PPAR)-a/g agonist, after aorta clamping in rats. Life Sci. 2022;297:120468.
- [CrossRef] [PubMed] [Google Scholar]




