


Translate this page into:
Vaspin inhibits ferroptosis: A new hope for treating myocardial ischemia–reperfusion injury

*Corresponding author: Duo Chen, Department of Special Inspection, The Second Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, Shandong, China. 15098865178@163.com
-
Received: ,
Accepted: ,
How to cite this article: Lin X, Xin L, Meng X, Chen D. Vaspin inhibits ferroptosis: A new hope for treating myocardial ischemia–reperfusion injury. CytoJournal. 2024;21:64. doi: 10.25259/Cytojournal_141_2024
Abstract
Objective:
Myocardial ischemia–reperfusion injury (MIRI) is a critical pathological basis for cardiovascular diseases. In recent years, the effect of ferroptosis on MIRI has attracted extensive attention. Vaspin, an adipose tissue-derived serine protease inhibitor, has multiple biological functions, including anti-inflammatory and antioxidant effects. This study aims to investigate the molecular mechanism by which vaspin alleviates MIRI by regulating hypoxia-inducible factor-1α (HIF-1α) and ferroptosis signaling pathways.
Material and Methods:
A mouse model of myocardial ischemia/reperfusion (I/R) and a hypoxia/reoxygenation (H/R) model was used to evaluate the protective effects of vaspin on MIRI. The mechanism by which ferroptosis is modulated by the vaspin/HIF-1α signaling pathway was investigated by constructing a vaspin overexpression adenoviral vector. Myocardial infarct size and histological changes were assessed using triphenyltetrazolium chloride and hematoxylin–eosin staining. Ferroptosis-related proteins were detected by Western blot assay, and apoptosis and reactive oxygen species levels were analyzed by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling. Iron content in myocardial tissue and cells was measured by enzyme-linked immunosorbent assay.
Results:
Myocardial I/R increased myocardial infarct size and serum lactate dehydrogenase (LDH) levels compared with the control group, indicating severe myocardial injury. Western blot results showed that MIRI reduced endogenous vaspin and HIF-1α levels and inhibited glutathione peroxidase 4. In vivo and in vitro vaspin overexpression treatment reduced infarct size, decreased LDH levels, inhibited ferroptosis pathway activity, and alleviated oxidative stress levels in myocardial tissues. In the H/R model, vaspin overexpression upregulated HIF-1α, inhibited ferroptosis markers, and reduced apoptosis and iron deposition. However, inhibiting HIF-1α reversed the cardioprotective and anti-ferroptotic effects of vaspin.
Conclusion:
Vaspin inhibits ferroptosis and upregulates the HIF-1α signaling pathway to mitigate myocardial I/R injury. The vaspin/HIF-1α pathway could be a potential target for MIRI prevention and treatment and offers fresh perspectives on ischemic heart disease management. Vaspin could be a novel cardioprotective agent that plays a significant role in the prevention and treatment of cardiovascular diseases.
Keywords
Vaspin
hypoxia-inducible factor-1α
Ferroptosis

INTRODUCTION
Myocardial ischemia–reperfusion injury (MIRI) is a considerable research focus in contemporary cardiology.[1,2] When reperfusion is applied to ischemic myocardium, the restoration of blood flow can alleviate local hypoxia but also trigger a series of complex pathophysiological changes that exacerbate myocardial cell injury.[3] The related pathological mechanisms include oxidative stress, calcium overload, inflammatory response, and programmed cell death. Ferroptosis, a novel form of cell death, has gained significant research attention. Ferroptosis primarily involves intracellular iron metabolism and accumulation of lipid peroxides.[4] In contrast to apoptosis and necrosis, ferroptosis displays unique cellular morphology and biochemical characteristics.[5,6] Understanding and controlling this process are crucial to mitigate the effects of MIRI.[7] However, protective strategies targeting ferroptosis remain in the research phase, and precise regulatory mechanisms are not fully elucidated.[8]
Vaspin (visceral adipose tissue-derived serine protease inhibitor) holds potential for improving metabolic syndrome and related cardiovascular diseases.[9,10] Visceral adipose tissue secretes vaspin, a serine protease inhibitor that was first found in obese people and is linked to increased insulin sensitivity. Vaspin exhibits anti-inflammatory, antioxidant, and anti-apoptotic effects.[11] In experimental MIRI models, vaspin can reduce myocardial cell death and improve cardiac function, although its specific mechanisms remain unclear.[12,13] Hypoxia-inducible factor-1α (HIF-1α), a crucial regulator, activates several downstream genes related to glycolysis, angiogenesis, and iron metabolism, thereby allowing cells to adapt to hypoxic conditions.[14] HIF-1α has low levels of oxidative stress and apoptosis in MIRI models.[15,16] HIF-1α can inhibit ferroptosis by downregulating hepcidin and ferritin.[16,17] Therefore, HIF-1α may be a crucial pathway through which vaspin ameliorates MIRI. This study reviews relevant literature to explore the mechanisms by which vaspin functions in MIRI.
Despite current research on the potential roles of vaspin and HIF-1α in cardiovascular protection, the mechanism through which vaspin inhibits ferroptosis through the regulation of HIF-1α to alleviate MIRI remains unclear. The direct regulatory effects of vaspin on HIF-1α expression have not been comprehensively studied. The specific effects of vaspin/HIF-1α axis on ferroptosis-related metabolic pathways should be investigated. Addressing these topics is crucial for understanding the role of vaspin in cardiovascular protection, in addition, whether vaspin can synergistically inhibit ferroptosis by modulating other signaling pathways, such as nuclear respiratory factor 2 (Nrf2) pathway, should be explored.[18] Nrf2 is a key transcription factor that regulates antioxidant stress responses and maintains iron homeostasis in cells by interacting with HIF-1α to protect cells from oxidative damage and iron overload.[19] Therefore, elucidating these complex interactions will help comprehensively understand the mechanisms of vaspin in MIRI.
This study aims to investigate the precise mechanisms by which vaspin reduces myocardial ischemia/reperfusion (I/R) injury by upregulating HIF-1α to prevent ferroptosis. We hypothesize that vaspin can activate the HIF-1α signaling pathway to regulate ferroptosis-related genes to reduce oxidative stress and lipid peroxidation damage in myocardial cells. The cardioprotective benefits of vaspin will be assessed using an in vitro hypoxia/reoxygenation (H/R) paradigm and an in vitro mouse model of MIRI. At the biological level, we will use gene knockout and pharmacological inhibition methods to verify the critical role of HIF-1α in the anti-ferroptosis process of vaspin and investigate the specific regulatory mechanisms of the vaspin/HIF-1α/Nrf2 signaling pathway in myocardial I/R injury.
This study, for the first time, will deeply elucidate the intrinsic mechanisms by which vaspin inhibits ferroptosis through the upregulation of HIF-1α and explore its protective effects on myocardial I/R injury. The innovation of this research lies in the systematic analysis of the relationships among vaspin, HIF-1α, and ferroptosis, providing new molecular targets and therapeutic strategies for myocardial I/R injury. Clarifying these mechanisms will not only enhance our understanding of the regulation of ferroptosis but also potentially provide a theoretical foundation and experimental basis for the clinical prevention and treatment of cardiovascular diseases. Results will open new avenues for the treatment of MIRI and advance cardiovascular disease research. The research findings could offer valuable insights for other diseases involving ferroptosis and thus present broad application prospects.
MATERIAL AND METHODS
Myocardial I/R model
Seventy-two specific pathogen free (SPF)-grade C57BL/6J mice (6–7 weeks old, weighing 18–22 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Production License No.: SYXK (Beijing) 2022-0052). The mice were acclimatized under 12-h light/dark cycle for 7 days and given free access to food and water. Food was withheld 12 h before modeling. Adeno-associated virus (AAV) vectors carrying OE-vaspin or OE-NC (negative control to OEvaspin) were constructed. The mice were injected through the tail vein with AAV vectors (2 × 1012 vector particles per mouse) carrying OE-vaspin or OE-NC for 3 consecutive days. The sham and MIRI groups were injected with saline through the tail vein. Five days after the injection of the AAV vectors, the mice underwent I/R surgery to establish an MIRI model. Plasmids were designed and synthesized by GenePharma Co., Ltd. (Shanghai, China). The MIRI model was induced by ligating left anterior descending artery (LAD) methods.[20,21] The mice were anesthetized with a 1% sodium pentobarbital solution (50 mg/kg) through intraperitoneal injection. After tracheal intubation, they were connected to a ventilator and fixed on a circulation-heated surgery table. Standard lead electrocardiograms were recorded. The surgical area was shaved and disinfected, and the heart was exposed through the third and fourth left intercostal spaces. Under a microscope (DM8000 M, Zeiss, Oberkochen, Baden-Württemberg, Germany), the LAD was sutured with a 7-0 silk suture, 2–3 mm below the left atrial appendage. Two loops approximately 5 cm long were placed at both ends of the ligature and tightened, which caused the anterolateral left ventricular myocardium to turn pale. A persistent ST-segment elevation on the electrocardiogram indicated successful ligation. The two loops were positioned at either side of the incision and left outside the body, and the chest was closed in layers. After 60 min of ischemia, the loops were gently pulled out horizontally from both sides of the mouse’s body simultaneously. A partial reduction in ST-segment elevation indicated successful reperfusion. The sham group was not subjected to LAD ligation during thoracotomy. After all the procedures, the mice were killed by neck dislocation.
In the erastin treatment group, I/R mice were treated with erastin (HY-15763, MedChemexpress, Monmouth County, NJ, USA) for 20 days at 10 μM/day. In the oltipraz treatment group, the mice were treated with 75 mg/kg/d oltipraz for 4 days.[22]
Triphenyltetrazolium chloride (TTC) staining and infarction size measurement
The heart was removed and rinsed in saline. The right ventricle was excised, and the heart was frozen on dry ice for 15 min. The heart was then sectioned into slices approximately 1 mm thick along the long axis of the left ventricle. The slices were incubated in 1.5% TTC staining solution (TTC, G3005, Solarbio, Beijing, China) in a 37°C incubator for 30 min in the dark. After staining, the slices were washed in phosphate-buffered saline (PBS) and then fixed in 4% paraformaldehyde for 30 min before being photographed. Image analysis was performed using ImageJ software. The red and white regions represented the area at risk (AAR), the white areas indicated infarcted myocardium (IA), and the red areas represented non-infarcted ischemic myocardium. The proportion of AAR to the left ventricle was used to compute the region of myocardial ischemia, and the percentage of IA to AAR was used to calculate the size of the infarct.
Hematoxylin-eosin (HE) staining to assess histopathological changes in myocardial tissue
After removal, a part of the myocardium was stored at −80°C, and another piece was embedded in paraffin after being fixed for 24 h in 4% paraformaldehyde (P1110, Solarbio, Beijing, China) to obtain continuous coronary 4 μm-thick slices. The specimen was deparaffinized in xylene and stained with hematoxylin for 4 min and eosin for 2 min using an HE staining kit (G1120, Solarbio, Beijing, China). Histopathological changes in the myocardial tissue were observed under a light microscope, and images were captured using a digital camera attached to the microscope.
Western blot analysis
Total proteins were isolated from heart tissue and heart cells. Protein concentrations were determined using a bicinchoninic acid assay (BCA) Protein Assay Kit (23225, Thermo Fisher Scientific, Waltham, MA, USA). Using sodium dodecyl sulfate polyacrylamide gel electrophoresis, the protein are deposited onto membranes. Electrophoresis was performed using a PowerPace Basic Electrophoresis System (1656019, Bio-Rad, Hercules, CA, USA). Following membrane blocking, the membranes were added with primary antibodies (1:1000) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:2000) and incubated for an additional night at 4°C. Goat serum working solution was provided by Beijing Zhongshan jinqiao Biotechnology Co., Ltd. After washing the membranes three times, they were treated with secondary anti-rabbit immunoglobulin (Ig) G (horseradish peroxidase) antibodies (1:2000) for 1 h at room temperature. Goat anti-rabbit IgG (H+L) secondary antibodies (ab6721) were used. The immune complexes on the membranes were visualized using enhanced chemiluminescence (ECL) (BL520B, Biosharp Life Science, Hefei, Anhui, China) and imaged with an E-Gel Imager (G8100, Invitrogen, Waltham, MA, USA). Grayscale analysis was conducted using ImageJ software (v1.8.0.345, National Institutes of Health, Bethesda, MD, USA) to quantify target protein expression. Primary and secondary antibodies were purchased from Abcam, Cambridge, MA, USA.
The primary antibodies were VASPIN (ab267470), HIF1a (ab92498), Nrf2 (ab62352), glutathione peroxidase 4 (GPx4) (ab125066), ferritin heavy chain 1 (FTH1) (ab183781), and GAPDH (ab9485).
Enzyme-linked immunosorbent assay (ELISA) detection of iron ions, GPx4, lactate dehydrogenase (LDH), malondialdehyde (MDA), and glutathione (GSH)
Ferrous Ion (Fe2+) Detection Kit (E-BC-K304-S) was obtained from EIAab Science Co., Ltd., Hubei, China. The mouse LDH ELISA Kit (ml002267) was purchased from Enzyme-linked Biotechnology Co., Ltd. (Shanghai, China). The mouse GPx4 ELISA Kit (CB10974-Mu) was obtained from COIBO BIO, Shanghai, China. The Lipid Peroxidation MDA Assay Kit (S0131S) was provided by Beyotime Biotechnology (Shanghai, China). Standard samples from the ELISA kits were diluted to different concentrations to create standard curves. Absorbance was measured by enzyme-labeled instrument (Varioskan LUX, Thermo Fisher Scientific, Waltham, MA, USA). The levels of iron ions, LDH, MDA, and GSH in the samples were determined based on their absorbance values and the standard curves.
Cell culture and transfection
Human cardiomyocytes (HCM cells, BFN60808575) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were mycoplasma-free, and the short tandem repeat (STR) analysis revealed that they were derived from the parental cells. The cardiomyocytes were cultivated in a high-glucose medium supplemented with 1% penicillin/streptomycin (P7630, Solarbio, Beijing, China) and 10% fetal bovine serum (S9020, Solarbio, Beijing, China) at 37°C under normoxic conditions (21% O2 and 5% CO2). The culture medium was refreshed every 24–48 h after washing with D-Hank’s solution 2–3 times. When cell confluence reached 70–80%, the cells were passaged using 0.25% trypsin digestion. The adherent myocardium cells were divided into two groups to investigate the effects of vaspin overexpression on H/R-treated cardiomyocytes: H/R + Empty Vector Group and H/R + Vaspin Overexpression Group. The cells were transfected with a suitable titer of empty vector adenovirus or vaspin-overexpressing adenovirus for 48 h. Lipofectamine™ 2000 transfection reagent was purchased from Life Technologies (12566014, Rockville, MD, USA). The cells were then cultured in glucose-free, serum-free Dulbecco’s Modified Eagle Medium (DMEM) medium for 8 h (5% CO2), returned to the complete medium, and cultured again for 12 h under normal conditions (5% CO2, 95% air, 37°C).
Establishment of H/R model
Cardiomyocytes were cultured in complete medium. After the cells adhered, the medium was replaced with glucose-free, serum-free DMEM and incubated for 8 h (5% CO2, 95% N2). The medium was changed back to the original complete medium, and the cells were further incubated under normal conditions (5% CO2, 95% air, 37°C) for 12 h.
Detection of reactive oxygen species (ROS) levels in cardiomyocytes by dihydroethidium (DHE) fluorescence staining
The ROS levels in cardiomyocytes were detected using the DHE fluorescent probe. The cells were incubated with 10 μmol/L DHE in a dark chamber at 37°C for 20 min and then washed 3 times with PBS. The DHE fluorescent probe (C400) was purchased from Invitrogen (Waltham, MA, USA). The fluorescence intensity of the DHE staining was measured using a fluorescent microscope, and Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA) was used to analyze every image. For quantification, three random fields were selected from each section, and the average fluorescence intensity of positively stained cells was analyzed.
Cell counting kit-8 (CCK-8) assay
HCM cells were seeded into 96-well plates at 5000 cells per well, with five replicates for each group. The cells were cultured for 24 h at 37°C with 5% CO2 to enable adhesion. According to the experimental design, the cells were divided into different groups: control, H/R, H/R + vaspin, and H/R + vaspin + HIF-1α inhibitor, with each group having five replicates. Appropriate treatments, including H/R, vaspin adenovirus transfection, and HIF-1α inhibitor, were applied for 24 h. Each well was added with 100 μL of serum-free medium containing 10% CCK-8 reagent and incubated in the dark for 2 h. The CCK-8 kit (C0037) was purchased from Beyotime Biotechnology Co., Ltd. (Shanghai, China). After incubation, the optical density value at 450 nm was measured using a microplate reader (Varioskan LUX, Thermo Fisher Scientific, Waltham, MA, USA).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
The treated cardiomyocytes were incubated with Proteinase K solution (2 g/L) diluted in PBS to a final concentration of 20 mg/L. The TUNEL assay kit (T2196) was purchased from Solarbio Science and Technology Co., Ltd. (Beijing, China). Each coverslip was incubated with 100 μL of Proteinase K solution at room temperature for 20 min. Following this, ×5 equilibration buffer was diluted with deionized water in a 1:5 ratio (20 μL of ×5 equilibration buffer + 80 μL deionized water) to make a ×1 equilibration buffer. Each sample was treated with 100 μL of ×1 equilibration buffer at room temperature for 20 min. The bright red labeling mix was thawed on ice, and terminal deoxynucleotidyl transferase (TdT) solution was prepared. After removing the buffer, the TdT was added to the cells and incubated at 37°C for 60 min, and washed with PBS 3 times (each for 5 min). Subsequently, the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (1:1000, C0065, Solarbio, Beijing, China) for 5 min to label the nuclei. After completing the above steps, the coverslips were mounted.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 8.0 software (GraphPad Software, Inc., San Diego, CA, USA). The measurement data were expressed as mean ± standard deviation. A t-test was used for comparisons between two groups, while one-way analysis of variance was used for multiple group comparisons. A P < 0.05 was considered statistically significant.
RESULTS
Myocardial I/R induces ferroptosis and decreases vaspin expression levels
In this study, we first established a mouse model of myocardial I/R to explore the impacted of I/R injury on myocardial tissue and changes in vaspin expression. The TTC and HE staining results showed that the myocardial infarction area in the I/R group was larger than that in the sham operation group [Figure 1a-c], indicating that I/R caused significant myocardial damages. The serum LDH activity in the I/R group was higher than that in the sham operation group [Figure 1d], further confirming severe myocardial tissue damage. To investigate the role of vaspin in I/R-induced myocardial injury, we measured vaspin and ferroptosis-related proteins. The Western blot results showed that the levels of vaspin, HIF-1α, Nrf2, GPx4, and FTH1 in the myocardial tissue of the I/R group were lower than those in the sham operation group [Figure 1e and f], suggesting that I/R downregulated vaspin expression and induced the activation of the ferroptosis pathway. To further verify the role of ferroptosis in I/R myocardial injury, we measured myocardial tissue iron levels and key indicators of ferroptosis, including GPx4 activity and GSH levels. The myocardial tissue iron levels in the I/R group were higher than those in the sham operation group [Figure 1g], while the GPx4 activity and GSH levels were lower [Figure 1h and i], suggesting that I/R induced ferroptosis. These results suggest that vaspin may play a protective role in I/R myocardial injury, providing new insights and experimental evidence for further investigation into the cardioprotective mechanisms of vaspin.
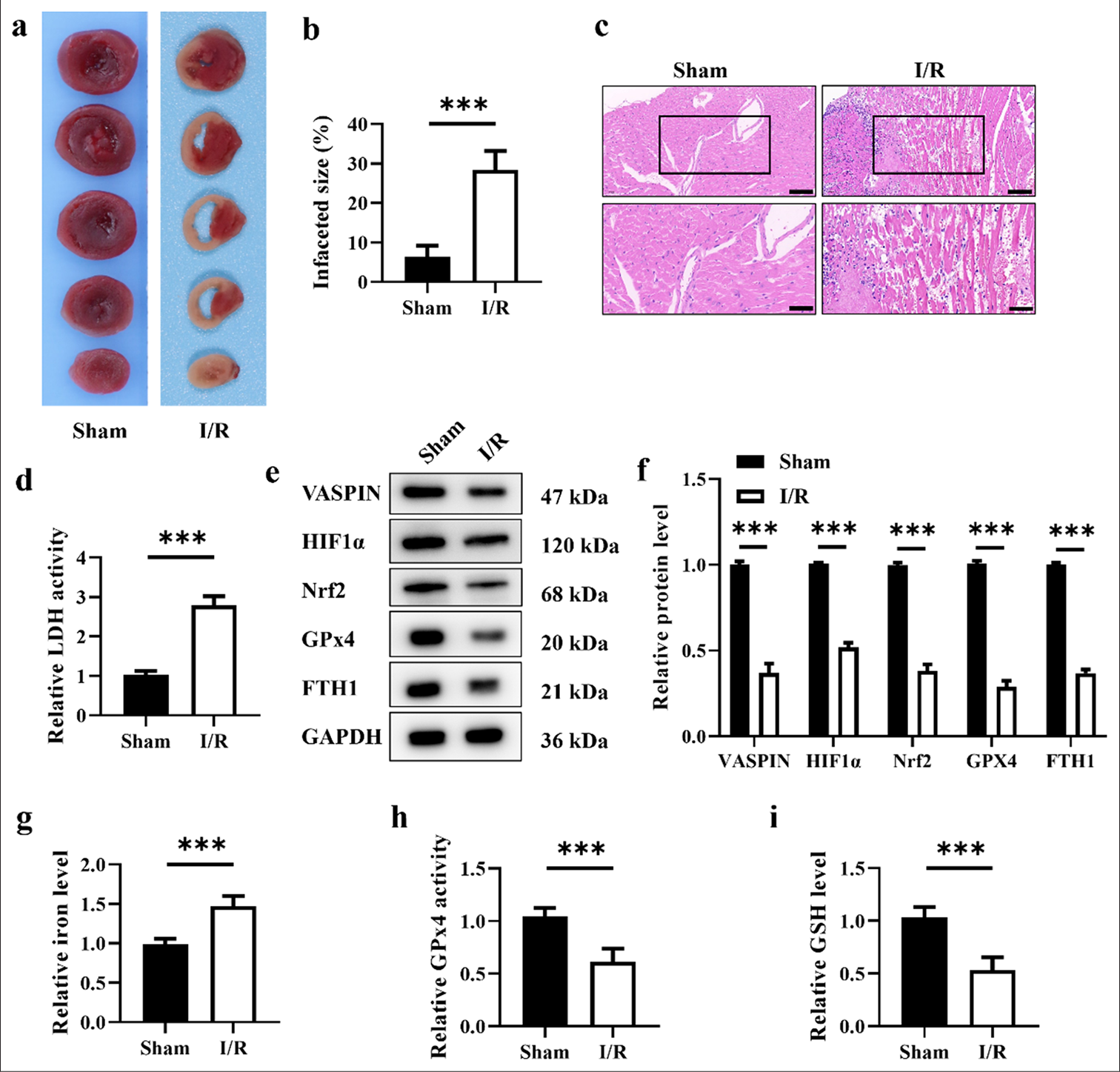
- Myocardial ischemia/reperfusion (I/R) induces ferroptosis and reduces the expression levels of vaspin. (a) Representative triphenyltetrazolium chloride staining images of myocardial sections. (b) Quantitative analysis of myocardial infarct size. (c) Hematoxylin-eosin staining of myocardial sections. Scale bar = 200 μm. Scale bar = 100 μm (enlarged view). (d) Relative lactate dehydrogenase activity in the sham and I/R groups. (e and f) Western blot analysis of vaspin, hypoxia-inducible factor-1α (HIF1α), nuclear respiratory factor 2 (Nrf2), glutathione peroxidase 4 (GPx4), and FTH1 in the sham and I/R groups. (g) Relative cellular iron levels. (h) Relative GPx4 activity in the sham and I/R groups. (i) Relative cellular glutathione levels. Results were presented as mean ± standard deviation. n = 6; ✶✶✶P < 0.001. FTH1: Ferritin heavy chain 1; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Overexpression of vaspin reduces H/R-induced myocardial I/R injury in mice
The role of vaspin in myocardial I/R injury was further investigated. We constructed an adenovirus vector for vaspin overexpression (OE-Vaspin) and introduced it into mice through tail vein injection to establish a vaspin overexpressing I/R mouse model. [Figure 2a-c] showed that the myocardial infarction area in the I/R+OE-vaspin group was smaller than those in the I/R and I/R+OE-NC groups, suggesting that vaspin overexpression had a significant protective effect against I/R-induced myocardial injury. The serum LDH activity in the I/R+OE-vaspin group was lower than those in the I/R and I/R+OE-NC groups [Figure 2d], further confirming the protective effect of vaspin overexpression on the damaged myocardium. To elucidate the molecular mechanism of the cardioprotective effect of vaspin, we measured the impact of vaspin overexpression on ferroptosis-related proteins induced by I/R. The Western blot results showed that the levels of vaspin, HIF-1α, Nrf2, GPx4, and FTH1 in the myocardial tissue of the I/R+OE-vaspin group were higher than those in the I/R and I/R+OE-NC groups [Figure 2e and f]. Vaspin overexpression upregulated HIF-1α and Nrf2 expression, thereby increasing GPx4 and FTH1 expression and inhibiting I/R-induced ferroptosis. The expression of vaspin, HIF-1α, Nrf2, GPx4, and FTH1 was determined by immunohistochemical staining of the myocardial tissue. Similar to the Western Blot results, the positive signals of vaspin, HIF-1α, Nrf2, GPx4, and FTH1 in the myocardial tissue of the I/R+ OE-Vaspin group were higher than those of I/R and I/R+ OE-NC group [Figure 2g]. To further verify the inhibitory effect of vaspin overexpression on I/R-induced ferroptosis, we measured myocardial tissue iron levels and key indicators of ferroptosis, including GPx4 activity and GSH levels. The myocardial tissue iron levels in the I/R+OE-vaspin group were lower than those in the I/R and I/R+OE-NC groups [Figure 2h], while GPx4 activity and GSH levels were higher [Figure 2i and j]. Vaspin overexpression reduced I/R-induced myocardial injury by inhibiting ferroptosis. Overall, vaspin overexpression can alleviate myocardial I/R injury by upregulating HIF-1α and Nrf2, thereby increasing GPx4 and FTH1 expression and inhibiting I/R-induced ferroptosis.
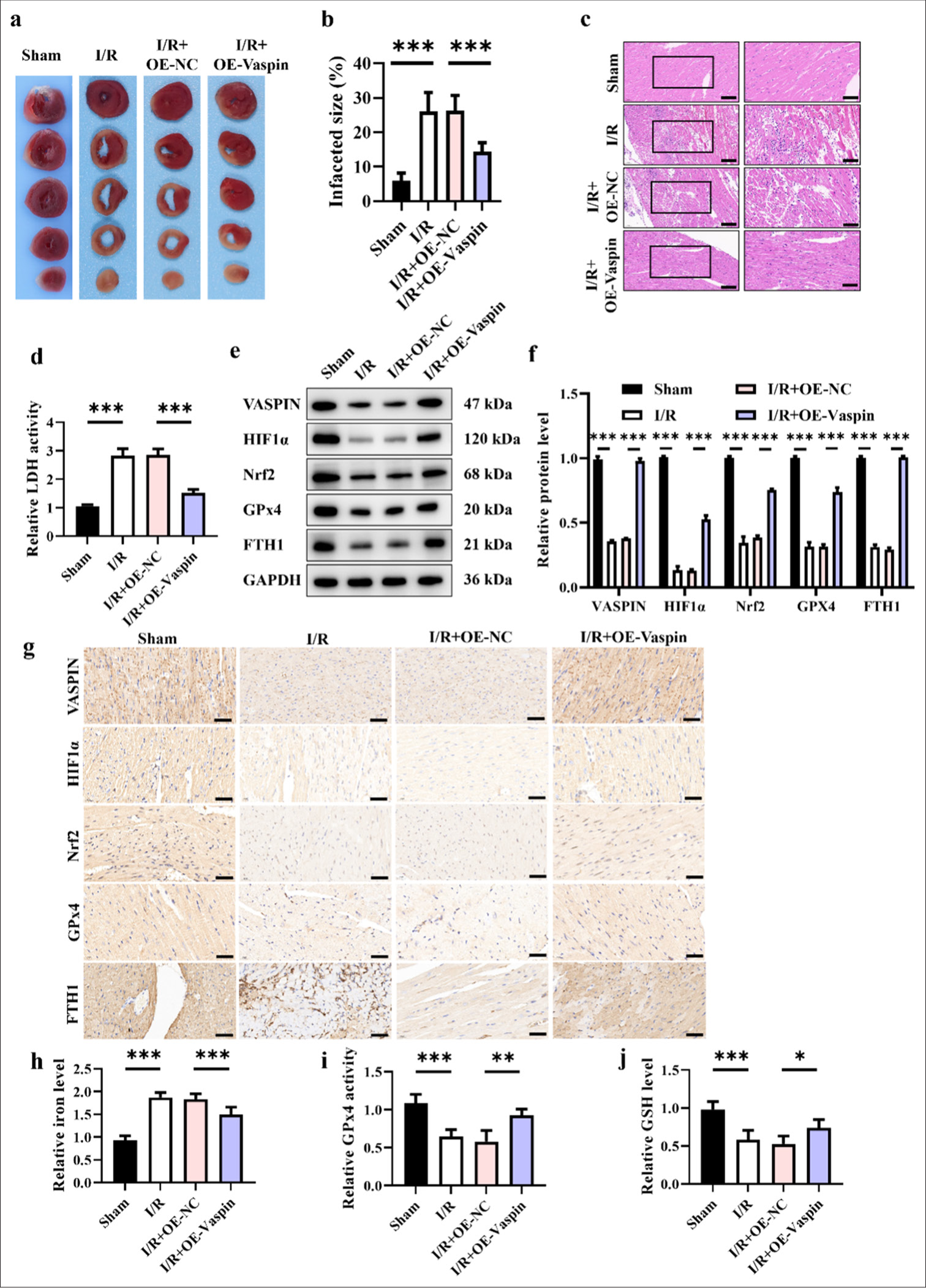
- Overexpression of vaspin reduces H/R-induced myocardial I/R injury in mice. (a) Representative triphenyltetrazolium chloride staining images. (b and c) Hematoxylin-eosin staining and quantitative analysis of myocardial infarct size. Scale bar = 200 μm. (d) Relative lactate dehydrogenase activity in each group. (e and f) Western blot analysis of Vaspin, hypoxia-inducible factor-1α (HIF1a), nuclear respiratory factor 2 (Nrf2), glutathione peroxidase 4 (GPx4), and FTH1. (g) Immunohistochemical analysis of Vaspin, HIF1a, Nrf2, GPx4, and FTH1. Scale bar = 200 μm. (h) Relative cellular iron levels. (i) Relative GPx4 activity. (j) Relative cellular glutathione levels. The results were presented as the mean ± standard deviation. n = 6; ✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001. OE-NC: Negative control to OE-vaspin, GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, FTH1: Ferritin heavy chain 1.
Overexpression of vaspin inhibits H/R-induced injury at the cellular level
We created an in vitro H/R model to replicate myocardial I/R to confirm the protective efficacy of vaspin against myocardial I/R injury. Initially, we overexpressed vaspin (OE-Vaspin) in HCM cardiomyocytes through adenoviral transduction to investigate its impact on H/R-induced cellular damage. [Figure 3a] shows that H/R treatment reduced cardiomyocyte viability, whereas the overexpression of vaspin markedly improved the decline in cell viability induced by H/R. In addition, the TUNEL staining indicated that H/R treatment increased cardiomyocyte apoptosis, while vaspin overexpression reduced H/R-induced apoptosis [Figure 3b and c]. These results imply that vaspin overexpression may mitigate cardiomyocyte damage and apoptosis caused by H/R. The levels of ROS and iron ion are critical indicators used to assess oxidative stress and ferroptosis in cells. We evaluated ROS levels in each group using fluorescent staining. H/R treatment increased ROS levels in cardiomyocytes, whereas overexpression of vaspin reduced H/R-induced ROS levels [Figure 3d and e]. We also measured iron ion levels in each group. The results indicated that H/R treatment elevated iron ion levels in cardiomyocytes while vaspin overexpression markedly decreased H/R-induced iron ion levels [Figure 3f]. These findings imply that Vaspin overexpression can mitigate H/R-induced oxidative stress and ferroptosis by reducing ROS and iron ion levels. GPx4 activity and GSH levels are important indicators for evaluating cellular damages and antioxidant capacity. Vaspin overexpression increased H/R-induced GPx4 activity in cardiomyocytes, and H/R treatment decreased GPx4 activity in these cells [Figure 3g]. Moreover, we assessed GSH levels in each group. The H/R treatment reduced GSH levels in cardiomyocytes, while vaspin overexpression elevated the decrease in GSH levels induced by H/R [Figure 3h]. Hence, vaspin overexpression can alleviate H/R-induced cellular damage and oxidative stress by increasing GSH levels and GPx4 activity.
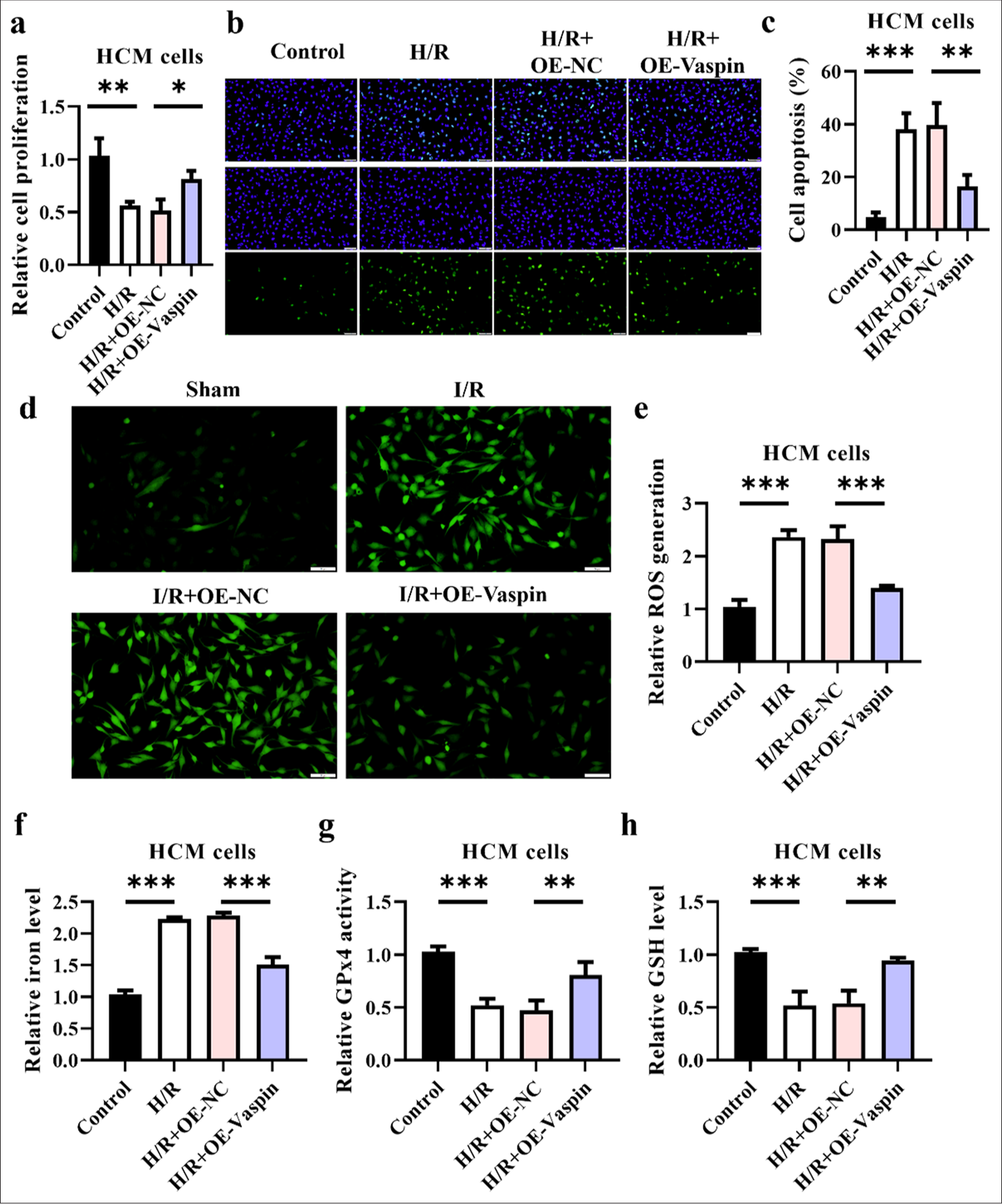
- Overexpression of vaspin inhibits hypoxia/reoxygenation (H/R)-induced damage at the cellular level. (a) Cell viability under H/R treatment was assessed by cell counting kit-8 assay in transfected cells. (b and c) Apoptosis under H/R treatment assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay in transfected cells. Scale bar = 200 μm. (d and e) Fluorescence staining to measure the relative cellular reactive oxygen species levels. Scale bar = 50 μm. (f) Relative cellular iron levels. (g) Glutathione peroxidase 4 activity in each group. (h) Glutathione levels. The results were presented as the mean ± standard deviation. n = 3; ✶✶P < 0.01, ✶✶✶P < 0.001.
Vaspin regulates H/R-induced ferroptosis
Figure 4 shows that vaspin regulates H/R-induced ferroptosis in cardiomyocytes. The Western blot results [Figure 4a and b] showed that H/R treatment decreased the levels of vaspin, HIF1α, Nrf2, GPx4, and FTH1, suggesting that H/R-induced ferroptosis in cardiomyocytes. The overexpression of vaspin could reverse the decrease in protein expression induced by H/R, with vaspin, Nrf2, and FTH1 restored to levels comparable with the control group. Although HIF1α and GPx4 expression was recovered, they remained lower than those in the control group. GPx4 is a crucial indicator for assessing ferroptosis. We further used immunofluorescence staining to detect the expression and localization of GPx4 in cardiomyocytes of each group [Figure 4c and d]. The GPx4 fluorescence intensity decreased following H/R treatment, whereas vaspin overexpression considerably enhanced GPx4 fluorescence signal, suggesting that vaspin inhibits H/R-induced ferroptosis by upregulating GPx4 expression.
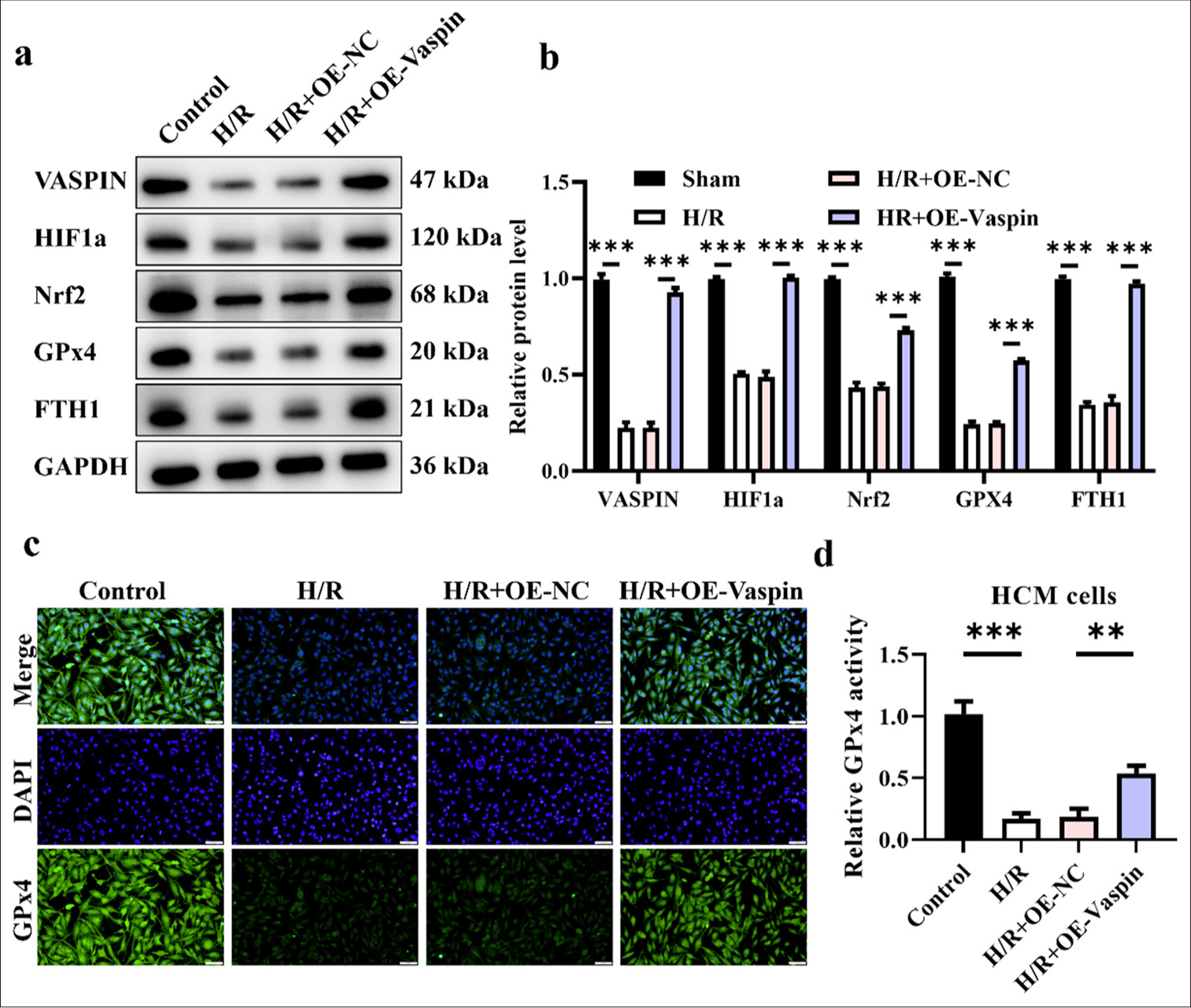
- Vaspin regulates hypoxia/reoxygenation (H/R)-induced ferroptosis. (a and b) Vaspin and ferroptosis-related protein expression levels in transfected cells after H/R treatment. (c and d) Immunofluorescence staining for the ferroptosis marker glutathione peroxidase 4 in myocardial cells. Scale bar = 100 μm. The results were presented as mean ± standard deviation. n = 3; ✶✶P < 0.01, ✶✶✶P < 0.001.
Ferroptosis inducer (Erastin) and HIF1a inhibitor (Oltipraz) reverse the cardioprotective effects of vaspin
We further validated that vaspin exerts its cardioprotective effects by upregulating HIF1a and inhibiting ferroptosis. The Western blot results [Figure 5a and b] showed that vaspin overexpression upregulated vaspin, HIF1a, Nrf2, GPx4, and FTH1 compared with the H/R group. However, treatment with the ferroptosis inducer erastin and the HIF1a inhibitor oltipraz reversed this upregulation of protein expression induced by vaspin. Vaspin possibly exerts its cardioprotective effects by inhibiting ferroptosis-related molecules through the upregulation of HIF1a. Cell viability assays [Figure 5c] indicated that vaspin overexpression increased the survival rate of cardiomyocytes following H/R treatment, while treatments with erastin and oltipraz reduced the cell survival rate to levels comparable with the H/R group. ROS is a major factor contributing to cellular injury. The fluorescence staining results [Figure 5d and e] demonstrated that vaspin overexpression reduced H/R-induced ROS levels, whereas treatments with erastin and oltipraz increased the ROS levels back to those observed in the H/R group. Elevated iron ion levels are a hallmark of ferroptosis. Vaspin overexpression reduced H/R-induced MDA levels, whereas erastin and oltipraz treatments raised MDA levels back to those observed in the H/R group in [Figure 5f]. We assessed the levels of free iron ions in each group [Figure 5g], and vaspin overexpression reduced H/R-induced iron ion levels. However, erastin and oltipraz treatments increased iron ion levels back to those observed in the H/R group. The GPx4 intensity [Figure 5h] indicated that vaspin overexpression increased H/R-induced GPx4 intensity, whereas erastin and oltipraz treatments reduced GPx4 intensity back to the levels observed in the H/R group. GSH level assays [Figure 5i] revealed that vaspin overexpression inhibited H/R-induced GSH depletion, while erastin and oltipraz treatments reduced GSH levels back to the levels in the H/R group. In summary, the ferroptosis inducer erastin and the HIF1a inhibitor oltipraz can reverse the protective effects of vaspin overexpression on H/R-induced cardiomyocyte injury.
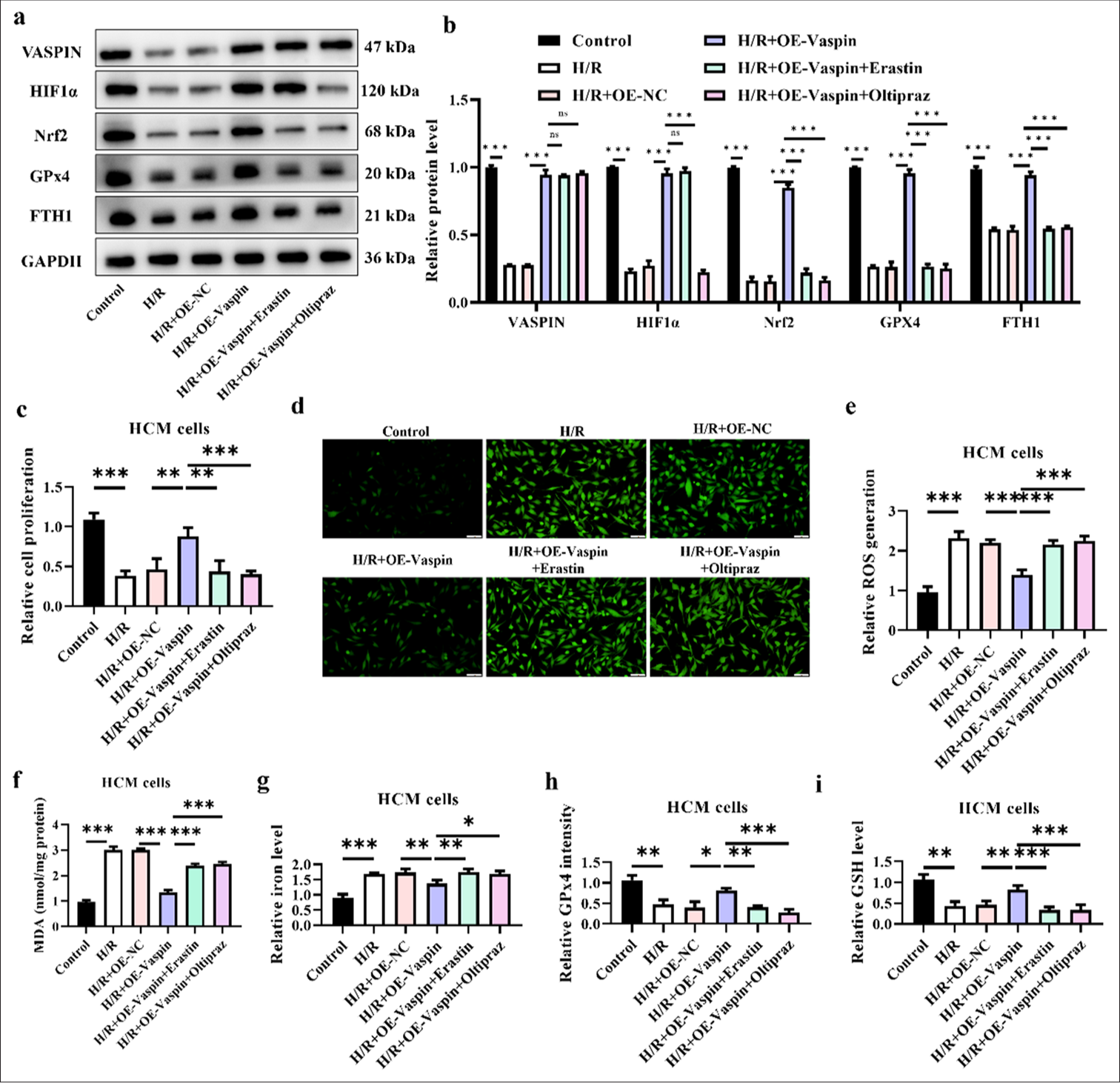
- Ferroptosis inducer (Erastin) and hypoxia-inducible factor-1α (HIF1α) inhibitor (Oltipraz) reverse the protective effects of vaspin on myocardium. Myocardial cells were pre-treated with Erastin (5 μM) or Oltipraz (10 μM) for 24 h before hypoxia/reoxygenation (H/R) treatment. (a and b) Vaspin and ferroptosis-related protein expression levels in transfected cells after H/R treatment. (c) Cell viability under H/R treatment was assessed by cell counting kit-8 assay in transfected cells. (d and e) Fluorescence staining to measure the relative cellular reactive oxygen species levels. Scale bar = 100 μm. (f) Relative malondialdehyde levels. (g) Relative cellular iron levels. (h) Glutathione peroxidase 4 intensity in each group. (i) Glutathione levels. The results were presented as mean ± standard deviation. n = 3; ✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001.
Animal models confirm that the ferroptosis inducer (Erastin) and HIF1a inhibitor (Oltipraz) reverse the cardioprotective effects of vaspin
We performed corresponding measures in a mouse model of myocardial I/R to further confirm the mechanism by which vaspin mitigates myocardial I/R injury through the activation of HIF1a and suppression of ferroptosis. The HE staining results [Figure 6a and b] indicated that vaspin overexpression reduced the myocardial infarction area compared with the I/R group. However, the ferroptosis inducer erastin and the HIF1a inhibitor oltipraz reversed the cardioprotective effects of vaspin and increased the infarct area to levels comparable with the I/R group. In [Figure 6c], vaspin overexpression reduced I/R-induced MDA levels, whereas erastin and oltipraz treatments increased the MDA levels back to those observed in the I/R group. Iron ion level assessments in myocardial tissue [Figure 6d] demonstrated that vaspin overexpression reduced the I/R-induced increase in the iron ion levels. However, erastin and oltipraz blocked this effect, increasing the iron ion levels back to those in the I/R group. The elevation of iron ion level is a critical feature of ferroptosis, suggesting that vaspin may exert cardioprotective effects by inhibiting ferroptosis. LDH release and GSH depletion are crucial indicators of myocardial injury and oxidative stress. GPx4 intensity assays [Figure 6e] showed that vaspin overexpression reduced I/R-induced GPx4 release, whereas erastin and oltipraz blocked this effect, thereby increasing GPx4 intensity back to those in the I/R group. GSH level assessments [Figure 6f] revealed that vaspin overexpression inhibited I/R-induced GSH depletion, while erastin and oltipraz reversed this effect, thereby reducing the GSH levels back to those in the I/R group. The animal experiments further confirmed that vaspin exerts its cardioprotective effects by upregulating HIF1a and inhibiting ferroptosis. The ferroptosis inducer erastin and the HIF1a inhibitor oltipraz can reverse these protective effects of vaspin. This finding suggests that vaspin may alleviate myocardial I/R injury by upregulating HIF1a and subsequently inhibiting the downstream ferroptosis pathway.
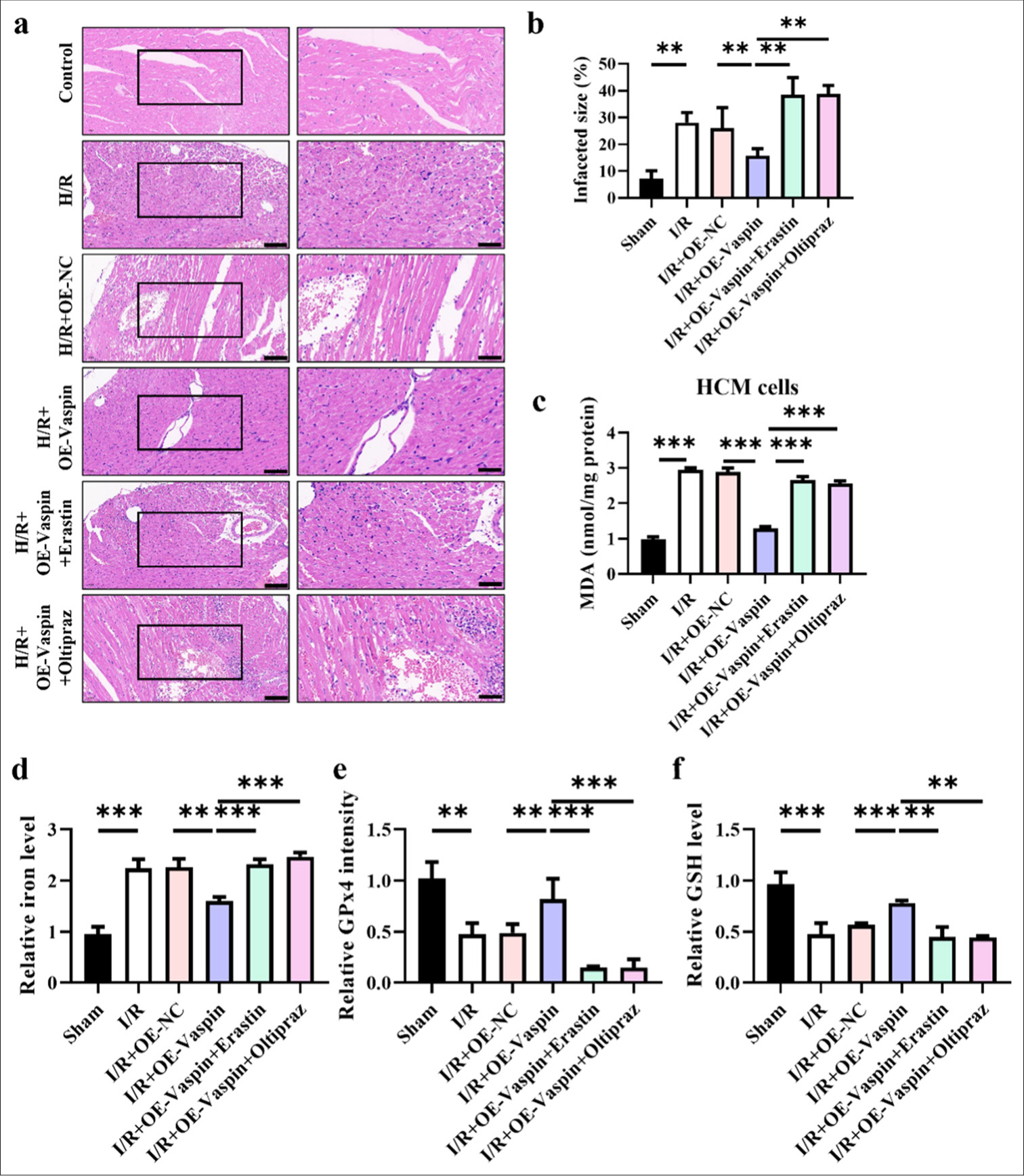
- Animal model confirms that ferroptosis inducer (erastin) and HIF1α (oltipraz) reverse the protective effects of Vaspin on myocardium. (a and b) Hematoxylin-eosin staining and quantitative analysis of myocardial infarct size, evaluated by. Scale bar = 200 μm. (c) Relative malondialdehyde levels. (d) Relative cellular iron levels in cardiac tissues. (e) glutathione peroxidase 4 intensity in cardiac tissues. (f) glutathione levels in cardiac tissues. The results were presented as mean ± standard deviation. n = 6; ✶✶P < 0.01, ✶✶✶P < 0.001.
DISCUSSION
This study reveals the mechanism by which vaspin alleviates MIRI by inhibiting ferroptosis through the upregulation of HIF-1α. Vaspin increased HIF-1α in the MIRI and in vitro H/R cardiomyocyte models by inhibiting ferroptosis-related molecular markers. This phenomenon, in turn, reduced myocardial cell damage and improved cardiac function. This finding not only enriches the biological functions of vaspin but also provides new possibilities for the clinical prevention and treatment of MIRI.
During myocardial I/R, although restoration of blood supply can ameliorate hypoxia, the subsequent reperfusion often leads to oxidative stress, calcium overload, and inflammatory responses, thereby aggravating myocardial injury.[23] Previous studies demonstrated that vaspin can alleviate cardiovascular damages through its anti-inflammatory, antioxidant, and anti-apoptotic effects.[10,12,13] Our study further confirms that vaspin, by upregulating HIF-1α expression, can reduce myocardial cell death and improve cardiac function in MIRI. Specifically, we systematically evaluated the protective effects of vaspin during myocardial I/R using animal and in vitro models. The results indicated a significant reduction in myocardial infarct size and improvement in cardiac function parameters in the vaspin-treated groups.
HIF-1α, a key adaptive factor under hypoxic conditions, plays an important role in regulating cell survival, metabolic reprogramming, and protecting tissues from I/R injury.[24]
Vaspin upregulated HIF-1α expression, but the mechanism remains unclear. We speculate that this may be related to the antioxidative stress function of vaspin. Specifically, vaspin might reduce the production of ROS, thereby stabilizing HIF-1α and allowing it to exert a sustained protective effect under hypoxic conditions. Another possible mechanism is that vaspin could influence iron metabolism by indirectly regulating HIF-1α stability. Iron metabolism is closely linked to HIF-1α degradation because iron-dependent hydroxylases promote HIF-1α degradation under normoxic conditions. Vaspin might interfere with this process to enhance HIF-1α stability.
Ferroptosis plays a significant pathological role in myocardial I/R injury by exacerbating cellular damage through increased oxidative stress and lipid peroxide accumulation.[25-27] Our study shows that vaspin reduces markers of ferroptosis, such as the upregulation of GPx4. These results indicate that vaspin mitigates myocardial cell damages post-I/R by inhibiting ferroptosis. We hypothesize that this process may be regulated by HIF-1α.
In this study, we have preliminarily constructed the relationship network of the vaspin/HIF-1α/ferroptosis signaling pathway. First, vaspin upregulates HIF-1α, which, as an important transcription factor, can activate a series of downstream genes, such as VEGF, which is involved in angiogenesis and improvement of myocardial blood supply. Second, HIF-1α regulates intracellular iron balance by downregulating hepcidin and ferritin, thereby reducing iron accumulation and inhibiting ferroptosis. In addition, HIF-1α can activate the Nrf2 signaling pathway to enhance the cell’s antioxidative capacity to further protect myocardial cells.[28-30] Through genetic knockout and pharmacological inhibition, this study verifies the critical role of HIF-1α in the anti-ferroptosis process of vaspin and explores the specific regulatory mechanisms of the vaspin/HIF-1α/Nrf2 signaling pathway in myocardial I/R injury.
Despite revealing the mechanism by which vaspin inhibits ferroptosis by upregulating HIF-1α to alleviate MIRI, several key issues remain to be explored. First, the specific molecular mechanism by which vaspin upregulates HIF-1α has not been fully elucidated. As such, further studies should explore its specific action pathways through molecular biology and biochemical methods. Second, the interaction between vaspin and other important signaling pathways (such as the inflammatory response pathway and apoptotic pathway) is an important research direction. Understanding these interactions will aid in comprehensive understanding of the multifaceted therapeutic potential of vaspin. Future research should focus on the clinical application potential of vaspin, such as enhancing its expression in myocardium through drug or gene therapy and evaluating its clinical efficacy and safety.
CONCLUSION
This study is the first to identify the molecular mechanism by which vaspin inhibits ferroptosis by upregulating HIF-1α to mitigate myocardial I/R injury. Our findings indicate that vaspin not only improves the survival and function of myocardial cells through its known anti-inflammatory, antioxidant, and anti-apoptotic effects but also through specific regulation of ferroptosis. This finding opens up new avenues for the clinical prevention and management of myocardial I/R injury and sheds light on the function of ferroptosis in cardiovascular disorders. Future research should continue to investigate the molecular mechanisms and clinical application potential of vaspin to develop new methods for the treatment of cardiovascular diseases.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study were available from the corresponding author on reasonable request.
ABBREVIATIONS
AAR – Area at risk
AAV – Adeno-associated virus
DHE – Dihydroethidium
GPx4 – Glutathione peroxidase 4
H/R – Hypoxia/reoxygenation
HE – Hematoxylin-eosin
HIF-1α – Hypoxia-inducible factor-1α
I/R – Ischemia/reperfusion
IA – Infarcted myocardium
LDH – Lactate dehydrogenase
MIRI – Myocardial ischemia–reperfusion injury
Nrf2 – Nuclear respiratory factor 2
ROS – Reactive oxygen species
SDS-PAGE – Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
TTC – Triphenyltetrazolium chloride
TUNEL – Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
AUTHOR CONTRIBUTIONS
XQM and DC: Designed the study; all authors conducted the study; LYX: Collected and analyzed the data; XHL: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, take public responsibility for appropriate portions of the content, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work are appropriately investigated and resolved.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study has been approved by the ethics committee of the Affiliated Hospital of Shandong University of Traditional Chinese Medicine, approval No. 2024017-KY. The informed consent is not required as there are no humans involved in this article.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
Not applicable.
References
- Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc Med. 2023;33:357-66.
- [CrossRef] [PubMed] [Google Scholar]
- Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17:773-89.
- [CrossRef] [PubMed] [Google Scholar]
- Reactive oxygen species-based nanomaterials for the treatment of myocardial ischemia reperfusion injuries. Bioact Mater. 2022;7:47-72.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis: Opportunities and challenges in myocardial ischemia-reperfusion injury. Oxid Med Cell Longev. 2021;2021:9929687.
- [CrossRef] [Google Scholar]
- Role of epigenetic regulation in myocardial ischemia/reperfusion injury. Pharmacol Res. 2021;170:105743.
- [CrossRef] [PubMed] [Google Scholar]
- Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. 2022;808:145968.
- [CrossRef] [PubMed] [Google Scholar]
- Myocardial ischemia-reperfusion injury: Therapeutics from a mitochondria-centric perspective. Cardiology. 2021;146:781-92.
- [CrossRef] [PubMed] [Google Scholar]
- Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin Pharmacother. 2020;21:1851-65.
- [CrossRef] [PubMed] [Google Scholar]
- New insights into cell apoptosis and proliferation: The potential role of vaspin. J Physiol Pharmacol. 2021;72:831-844.
- [Google Scholar]
- Effect of vaspin on myocardial ischemia-reperfusion injury rats and expression of Nlr family pyrin domain containing 3 (nlrp3) J Biomater Tissue Eng. 2020;10:895-900.
- [CrossRef] [Google Scholar]
- Visceral adipose tissue-derived serine protease inhibitor: A unique insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci U S A. 2005;102:10610-5.
- [CrossRef] [PubMed] [Google Scholar]
- Vaspin protects rats against myocardial ischemia/reperfusion injury (MIRI) through the TLR4/NF-κB signaling pathway. Eur J Pharmacol. 2018;835:132-9.
- [CrossRef] [PubMed] [Google Scholar]
- Nesfatin-1 and vaspin as potential novel biomarkers for the prediction and early diagnosis of gestational diabetes mellitus. Int J Mol Sci. 2019;20:159.
- [CrossRef] [PubMed] [Google Scholar]
- Diazoxide post-conditioning activates the HIF-1/HRE pathway to induce myocardial protection in hypoxic/reoxygenated cardiomyocytes. Front Cardiovasc Med. 2021;8:711465.
- [CrossRef] [PubMed] [Google Scholar]
- Ozone preconditioning protects rabbit heart against global ischemiareperfusion injury in vitro by up-regulating HIF-1α. Biomed Pharmacother. 2022;150:113033.
- [CrossRef] [PubMed] [Google Scholar]
- FOXN4 affects myocardial ischemia-reperfusion injury through HIF-1α/MMP2-mediated ferroptosis of cardiomyocytes. Cell Mol Biol (Noisy-le-grand). 2023;69:214-25.
- [CrossRef] [Google Scholar]
- Bioinformatics integration reveals key genes associated with mitophagy in myocardial ischemia-reperfusion injury. BMC Cardiovasc Disord. 2024;24:183.
- [CrossRef] [PubMed] [Google Scholar]
- Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int J Biol Macromol. 2019;125:496-502.
- [CrossRef] [PubMed] [Google Scholar]
- HIF1, HSF1, and NRF2: Oxidant-responsive trio raising cellular defenses and engaging immune system. Chem Res Toxicol. 2022;35:1690-700.
- [CrossRef] [PubMed] [Google Scholar]
- Glutamatergic neurons in lateral hypothalamus play a vital role in acupuncture preconditioning to alleviate MIRI. J Neurophysiol. 2023;129:320-32.
- [CrossRef] [PubMed] [Google Scholar]
- Piperine protects against pyroptosis in myocardial ischaemia/reperfusion injury by regulating the miR-383/RP105/AKT signalling pathway. J Cell Mol Med. 2021;25:244-58.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of Nrf2 in the liver is associated with stress resistance mediated by suppression of the growth hormone-regulated STAT5b transcription factor. PLoS One. 2018;13:e0200004.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting calcium homeostasis in myocardial ischemia/reperfusion injury: An overview of regulatory mechanisms and therapeutic reagents. Front Pharmacol. 2020;11:872.
- [CrossRef] [PubMed] [Google Scholar]
- HIF1α in myocardial ischemiareperfusion injury. Mol Med Rep. 2021;23:1-9.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting ferroptosis: Pathological mechanism and treatment of ischemia-reperfusion injury. Oxid Med Cell Longev. 2021;2021:1587922.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39:210-25.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021;7:193.
- [CrossRef] [PubMed] [Google Scholar]
- Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019;24:101210.
- [CrossRef] [PubMed] [Google Scholar]
- Andrographolide inhibits hypoxia-induced HIF-1α-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases 2/3 in human endothelial cells. Environ Toxicol. 2017;32:918-30.
- [CrossRef] [PubMed] [Google Scholar]
- Strange bedfellows: Nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants (Basel). 2017;6:27.
- [CrossRef] [PubMed] [Google Scholar]




