


Translate this page into:
Adenosine deaminase acting on RNA 2 provides neuroprotection by activating Kv1.1 channels in a rat epilepsy model


*Corresponding Author’s: Pengzhen Wang, Department of Traumatic Surgery, Guangzhou Red Cross Hospital, Jinan University, Guangzhou, China. wang521jnu@163.com
Bo Ning, Department of Neurosurgery, Guangzhou Red Cross Hospital, Jinan University, Guangzhou, China. ningbo1974@126.com
-
Received: ,
Accepted: ,
How to cite this article: Zhu P, Yuan W, Liu W, Wu J, Wang P, Ning B. Adenosine deaminase acting on RNA 2 provides neuroprotection by activating Kv1.1 channels in a rat epilepsy model. CytoJournal. 2024;21:57. doi: 10.25259/Cytojournal_53_2024
Abstract
Objective:
Potassium voltage-gated channel sub-family A member 1 (Kv1.1), as a shaker homolog potassium channel, displays a special mechanism for posttranscriptional regulation called RNA editing. Adenosine deaminase acting on RNA 2 (ADAR2) can cause abnormal editing or loss of normal editing, which results in cell damage and related diseases. The relationship between Kv1.1 and editing enzyme ADAR2 in epileptic rats remains incompletely understood. We aimed to investigate the neuroprotective role of ADAR2 and its relationship with Kv1.1 in epileptic rats and the SH-SY5Y neuroblastoma cell line.
Material and Methods:
A rat epilepsy model was induced in vivo using lithium chloride–pilocarpine. We investigated the effect of ADAR2 on epileptic rats through Western blotting, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and histological analysis. Western blotting was aimed at investigating the effect of overexpression of ADAR2 and Kv1.1-interfering RNA (si-Kv1.1) for neuronal apoptosis.
Results:
The overexpression of ADAR2 in epileptic rats led to the increased mRNA and protein expression of Kv1.1 (P < 0.001) and B-cell leukemia/lymphoma 2 protein (Bcl-2) (P < 0.001), whereas the decreased expressions of Bcl-2-associated X protein and cleaved caspases-3/7 at protein levels (P < 0.0001; P < 0.0001; P < 0.01) detected by Western blotting and qRT-PCR experiments. Hematoxylin and eosin staining and Nissl staining revealed the neuroprotection provided by ADAR2 overexpression. The experiments demonstrated that Kv1.1 was regulated by ADAR2. ADAR2 overexpression increased neuronal survival in in vivo experiments through the elevation of Bcl-2 levels (P < 0.05) and reduction of cleaved caspase-3/7 activity (P < 0.0001; P < 0.01). In the recovery experimental group that involved silencing Kv1.1, the beneficial effects of overexpressing ADAR2 were no longer observed (P < 0.05).
Conclusion:
Our findings confirm that the upregulation of ADAR2 promotes Kv1.1 protein expression, which ultimately reduces neuronal damage in the hippocampus of animals with epilepsy.
Keywords
Adenosine deaminase acting on RNA 2
Potassium voltage-gated channel subfamily A member 1
Epileptic
Neuron
Hippocampus

INTRODUCTION
Potassium channels perform a crucial role in neuronal electrical activity and are directly implicated in the pathogenesis of epileptic seizures.[1] Voltage-gated potassium ion currents in nerve cells are central to action potential repolarization in the central nervous system. A-type potassium channels are particularly important because they regulate multiple neuronal processes, such as neuronal excitability, signal transmission, cell differentiation, and apoptosis. These channels briefly open during depolarization, which generates transient outward currents known as A- type potassium currents (IA) currents.[2] The effect of numerous environmental toxins or drugs on neurons, whether protective or detrimental, hinges on their capability to modulate repolarization.[3] The amplitude of A-type potassium ion currents in Gamma-Aminobutyric Acid (GABAergic) neurons of the nucleus tractus solitarius exhibited a considerable reduction in a mouse epilepsy model. This reduction elevated neuronal excitability, which led to epilepsy and ultimately increased the risk of sudden death due to the condition.[4] Furthermore, in hypoxic newborn mice, the upregulation of A-type potassium channels inhibited neural excitability.[5] Therefore, alterations in the A-type current, which regulates neuronal excitability, may lead to abnormal neuron firing. However, further studies are needed to elucidate the specific subtypes of A-type potassium ion channels involved in this process and the precise underlying mechanisms.
In the initial stages of repolarization, Kv1 channels substantially influence the A-type potassium current, which is crucial for the maintenance of resting membrane potential stability, decelerated depolarization, and delaying action potential onset. Encoded by the KCNA1 gene, potassium voltage-gated channel subfamily A member 1 (Kv1.1) is the first cloned voltage-gated potassium channel in the mouse brain. This gene is widely expressed in the brainstem, hippocampus, cortex, and myelinated nerve fibers and exhibits tissue specificity.[6] Growing evidence associates Kv1.1 with neuronal excitability and apoptosis.[7] Mutations in KCNA1 lead to autosomal dominant episodic ataxia type 1, which is characterized by ataxia and myokymia. Furthermore, various phenotypes have been associated with this gene.[8,9] Mice lacking Kv1.1 channels display severe epilepsy or fatal seizures, which underscore the critical role of the Kv1.1 channel in neuronal excitability.[10] Kv1.1 shows promise as a target for the treatment of hyperexcitable brain disorders, and thus, further elucidation of its role in A-type potassium ion current modulation will improve our comprehension of the mechanisms underlying neuronal hyperexcitability and damage.
Researchers have been increasingly directing their attention toward comprehending the role of adenosine deaminase acting on RNA enzymes (ADARs), which influence chemical and electrical neurotransmission and voltage- and ligand-gated ion channels, and presynaptic release mechanisms.[11] In vertebrates, ADAR1 (ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2) encode proteins similar to ADAR, with ADAR1 and ADAR2 uniquely possessing deaminase catalytic activity, which affects editing stability and effectiveness.[12-15] ADAR3 is presumed to be catalytically inert. Various tissues express ADAR2, but the highest editing transcript levels can be found in the brain.[16] Enzymatic deamination by ADAR2 leads to an I400V substitution in Kv1.1 mRNA, which suggests a link between Kv1.1 dysregulation and epilepsy development.[17] Nevertheless, the research on the biological effect of ADARs and Kv1.1 on neurons is limited.
In an ideal scenario, epilepsy therapy should increase Kv1.1 activity to mitigate neural excitability and cellular damage. This study aimed to investigate the roles of ADAR2 and Kv1.1 in a rat epileptic model induced by lithium pilocarpine in the presence of chlorine to establish a theoretical framework for the advancement of epilepsy therapy.
MATERIAL AND METHODS
Experimental animals and epilepsy model induction
Male Sprague–Dawley rats of Specific Pathogen Free grade, weighing 200–250 g and aged 5–8 weeks, were obtained from the Guangdong Provincial Medical Laboratory Animal Center (license number: SCXK 2018-0002). The experiment was conducted with the approval of the Animal Ethics Committee of Guangzhou Red Cross Hospital (No. 2020-194-01, Date: January 09, 2020).
The rats were housed in a well-ventilated environment with a 12 h light-dark cycle, which maintained a constant temperature of 25°C and humidity of 50–60%. Throughout the rearing phase, the animals had unlimited access to food and water. Eighteen rats were randomly allocated to the sham, model, or Overexpression (oe)-ADAR2 groups. Induction of epilepsy in the model group was performed using a modified lithium chloride–pilocarpine method: the rats were injected with freshly prepared lithium chloride (BCCB9625, Sigma, MO,USA) solution (180 mg/kg), followed by an intraperitoneal injection of scopolamine ([1 mg/kg], CS-B0293, Shanghai C-reagent Biotechnology, Shanghai, China) 24 h later and a intraperitoneal injection of pilocarpine ([30 mg/kg], B20843, Shanghai Yuanye Bio-Technology, Shanghai, China) 30 min later.[18] If no seizures occurred within 30 min, additional pilocarpine hydrochloride (10 mg/kg) was administered intraperitoneally. The model group comprised rats with grades IV and V seizures. The control (Con) group received an injection of 0.9% sodium chloride solution. All efforts were exerted to minimize rat suffering and the number of utilized animals.
Adenovirus transfection
Adenovirus transfection was performed using adenovirus ADAR2 (Ad-ADAR2) to overexpress ADAR2. Intracerebroventricular injections were administered in accordance with established protocols.[19] The rats were anesthetized intraperitoneally using 0.3% sodium pentobarbital (P3761, Merck KGaA, USA). Rats were immobilized in a stereotactic frame, and a small burr hole was created in the skull at precise coordinates: 1.5 mm lateral to the sagittal suture and 0.8 mm posterior to the bregma. The left lateral ventricle was perforated using a 25-gauge needle, which was positioned 4.0 mm below the horizontal plane of the bregma. Six days before induction of the epilepsy model, 10 mL diluted Ad-ADAR2 was gradually injected into the lateral ventricle to ensure maximum amplification.
Establishment and intervention of in vitro epilepsy model
The SH-SY5Y neuroblastoma cell line was purchased from Shanghai Zhongqiao Xinzhou Biotechnology Co., Ltd (ZQ0050, Shanghai, China). Cell lines were authenticated by the Genomics Unit using short tandem repeat (STR) profiling (AmpFLSTR® Identifier® Plus polymerase chain reaction (PCR) Amplification Kit, 4427368, Thermo Fisher Scientific, MA, USA). STR analysis confirmed the match between the cell lines and expected profiles, which ensured their identity. Mycoplasma tests were performed on all cell lines every other week using the LycoAlert Mycoplasma Detection Kit (LT07-118, LONZA, BSL, Switzerland). All tests yielded negative results for mycoplasma contamination. SH-SY5Y cells were cultured in an incubator with 5% CO2 at 37 °C using Dulbecco’s Modified Eagle medium ([DMEM], 31600034, Gibco, GI, USA) supplemented with 10% fetal bovine serum ([FBS], 10099-141, Gibco, GI, USA) and 10% penicillin–streptomycin (15140-122,Gibco, GI, USA). The experimental groups included control (Con), EP (Epilepsy), oe-ADAR2, and recovery experimental groups (oe-ADAR2 + Kv1.1 interfering RNA [si-Kv1.1,GGAACG AGUACUUCUUCGA]).
In the Con group, cells were cultured in DMEM for 3 h followed by incubation in a fresh medium for 72 h. The model group included cells exposed to magnesium-free extracellular fluid at 37 °C in a 5% CO2 incubator for 3 h and then cultured in a 10% FBS DMEM medium for 72 h. The Mg2+-free extracellular solution contained (in mM) NaCl (145), KCl (2.5),N-(2-Hydroxyethyl)Piperazine-N’-(3-EthanesulfonicAcid) (10), CaCl2 (2), glucose (10), and glycine (0.002). Culturing of the oe-ADAR2 group was performed in magnesium-free extracellular fluid 3 h and DMEM medium containing Ad-ADAR2 solution in an incubator for 72 h. The recovery group cells subjected to 3 h incubation in magnesium-free extracellular fluid, followed by 72 h incubation in a DMEM medium containing AdADAR2 solution and si-Kv1.1. The results for the knockdown efficiency of kv1.1 was detected through quantitative reverse transcription PCR (qRT-PCR). Subsequently, cells were collected for Western blotting.
Histology
The rats were anesthetized intraperitoneally using 0.3% sodium pentobarbital and were executed after asphyxiation with high concentrations of carbon dioxide (CO2). Subsequently, their brain was collected, and the hippocampus was promptly isolated and perfused with 4% buffered formalin (1.00496, Sigma, MO, USA). Serial paraffin tissue sections were prepared for HE (C0105S, Shanghai Biyuntian Biological Co., Ltd, Shanghai, China) and Nissl staining in accordance with standard procedures, including dehydration, paraffin immersion, and embedding. Nissl staining was employed to assess the neuronal status in the rat hippocampal region, and immunohistochemical techniques were utilized to detect hippocampal neuronal apoptosis. For immunohistochemistry analysis, goat two-step detection kit was used to detect antigens in accordance with the manufacturer’s instructions (PV-8000, ZSGB-BIO, Beijing, China). Endogenous peroxidase activity was quenched using 3% (v/v) hydrogen peroxide. The sections were blocked with 3% (v/v) bovine serum albumin (BSA) and incubated with 100 mL diluted primary antibody of rabbit polyclonal anti-NeuN (24307, 1:1000, Cell Signaling Technology) overnight at 4°C and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. The reaction was visualized through incubation of the sections with a Diaminobenzidine kit (ZLI-9017, ZSGB-BIO, Beijing, China). Photographs were captured using an inverted microscope (BX61; Olympus, Tokyo, Japan). The percentage of neuron-positive cells in the total number of cells in a given field was calculated for all groups.
Immunofluorescence
Tissue sections were dewaxed and dehydrated, after which antigen retrieval was performed with 0.01 M citrate buffer (pH 6.0). The tissue sections were first blocked with 5% (v/v) BSA (SRE0096, Sigma, Mo, USA) for 1 h at 37°C, then incubated overnight at 4°C with the primary antibody against ADAR2 (1:1,000; sc-73409; Santa Cruz, CA, USA). Subsequently, the tissue sections were treated with a secondary fluorescein-conjugated (green) antibody (1:500; #4408; CST, BST, USA) solution for 1 h at 26°C. 4',6-Diamidino-2-phenylindole (DAPI) solution (C0060, Solarbio, Beijing, USA) was diluted in deionized water to a final concentration of 0.1 mg/mL. The slides were added with a few drops of DAPI solution, stained for 10 min, and washed thrice using deionized water. Images of stained cells were captured using a Zeiss LSM 510 META System CLSM at ×200 magnification and analyzed. The mean fluorescence intensity was analyzed using software ImageJ (Version:1.8.0.112,USA).
Western blotting
Chondrocytes (2 ×105 cells/well) were cultured in six-well plates and treated as described for immunofluorescence studies. Cells were lysed, and protein concentrations were determined through the BCA method. Proteins (20 mg/lane) were separated through 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (ISEQ00010, Merck Millipore, Darmstadt, German). The membranes were blocked with 5% BSA for 1 h at room temperature, and the proteins were sequentially probed with the corresponding primary antibodies overnight at 4°C.
Brains were promptly extracted from the euthanized animals, and their hippocampal structures were dissected. Before Western blotting, the cells were lysed in modified radioimmunoprecipitation assay buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-Cl [pH 8.0], and 0.1% sodium dodecyl sulfate) supplemented with protease and the phosphatase inhibitor phenylmethylsulfonyl fluoride (1 mM, 36978, Thermo Fisher Scientific, MA, USA). Following rapid homogenization, the homogenate was incubated in ice for 30 min and centrifuged at 12,000 g for 15 min at 4°C. Subsequently, proteins (30 mg/lane) were separated through 10% SDS-PAGE and transferred onto PVDF membranes (ISEQ00010, Merck Millipore). Following 1 h membrane blocking with 5% BSA at room temperature, the membranes were incubated overnight at 4°C with primary antibodies against Kv1.1 (1:1,000; abx134533, AmyJet Scientific Inc, Wuhan, China), ADAR2 (1:1,000; Santa Cruz, CA,USA), Bcl-2-associated X protein (Bax) (1:1,000; #2772, CST, BST, USA), B-cell leukemia/lymphoma 2 protein (Bcl-2) (1:1,000; #28150, CST, BST, USA), cleaved caspases 3 (1:1,000; #9664, CST, BST, USA), and 7 (1:1,000; #8438, CST, BST, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000; #5174, CST, BST, USA). Subsequently, the membranes were treated with an anti-rabbit/mouse Immunoglobulin G-HRP-linked antibody (1:3,000; 7074/7076, CST, BST, USA). Blots were detected using a chemiluminescence kit (GE2311, Genview, Beijing, China). The ChemiDoc XRS Imaging System (Bio-Rad, USA) was utilized for image acquisition, and Image Lab 5.2.1 software (Bio-Rad, USA) was employed for analysis.
qRT-PCR
Exactly 0.1 g brain tissue was homogenized in an ice bath with 1 mL Trizol reagent (15596026, Invitrogen, Thermo Fisher Scientific, MA, USA), and the cultured cells were lysed using 1 mL of the same reagent. The total RNA was purified and quantified using NanoDrop ND-1000 v3.3.0 (Thermo Fisher Scientific, Wilmington, DE, USA) following the manufacturer’s instructions. qRT-PCR was conducted in 96-well microplates using the All-in-One quantitative PCR Mix (GeneCopoeia, Rockville, MD, USA) labeled with Synergy Brands Green I dye. Reverse transcription was carried out using 1.0 mg total RNA using the PrimeScript® RT Master Mix (RR036A,TaKaRa, Shiga, Japan). The thermocycling conditions were as follows: Pre-denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 34 s, and annealing and extension at 55°C for 5 s. The housekeeping gene GAPDH was used to standardize the expression levels in each sample. The 2(−ΔΔCT) method was perform to ascertain the relative mRNA expression levels. Table 1 shows the primer sequences for all genes.
| Genes | Forward | Reverse |
|---|---|---|
| R-GAPDH | 5'-GGCACAGTCAAGGCTGAGAATG-3' | 5'-ATGGTGGTGAAGACGCCAGTA -3' |
| R-Kv1.1 | 5'-CCGCCGCAGCTCCTCTACTATCA-3' | 5'-CAAGGGTTTTGTTTGGGGGCTTTT-3' |
| R-ADAR2 | 5'-TGATAGACATCCGAATCGCAAAG-3' | 5'-TAGATGGGCTCCACGAAAATG-3' |
| H-GAPDH | 5'-GTCTCCTCTGACTTCAACAGCG-3' | 5'-ACCACCCTGTTGCTGTAGCCAA-3' |
| H-Kv1.1 | 5'-TAGTGCAGTGTACTTTGCCGA-3' | 5'-GTCACCGTATCCTACAGTGGT-3' |
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, Kv1.1: Potassium voltage-gated channel subfamily A member 1, ADAR2: Adenosine deaminase acting on RNA 2, A: Adenine, T: Thymine, G: Guanine, C: Cytosine; R: Rat, H: Human
Data analysis
The Statistical Package for the Social Sciences for Windows, Inc. (version 11.5; Chicago, IL, USA) was used in statistical analyses. Statistical analyses were performed, with the unpaired t-test used for two groups or one-way analysis of variance for multiple groups. Data were presented as mean ± standard error of the mean. All experiments were independently repeated thrice. The significance threshold was set at P < 0.05.
RESULTS
Effect of ADAR2 overexpression on Kv1.1 expression in the hippocampal subregion
Immunofluorescence analysis revealed the presence of ADAR2-positive cells (green) in the CA1 region of the hippocampus in the sham group [Figure 1a and b]. Conversely, a substantial reduction in ADAR2-positive neurons was observed in the model group (P < 0.001). In addition, the oe-ADAR2 group exhibited higher levels of ADAR2-positive cells in the CA1 region compared with the model group (P < 0.001). Western blot analysis confirmed the significant downregulation of ADAR2 and Kv1.1 protein expression in the model group compared with that in the sham group (ADAR2, P < 0.0001; Kv1.1, P < 0.001), which indicates the successful rat model development. Transfection of Ad-ADAR2 into model rats led to a significant increase in the protein expressions of ADAR2 and Kv1.1 [Figure 1c-e] (ADAR2, P < 0.001; Kv1.1, P < 0.0001). Changes in ADAR2 and Kv1.1 mRNA levels mirrored the trends in protein levels [Figure 1f and g] (ADAR2, P < 0.01; Kv1.1, P < 0.0001).
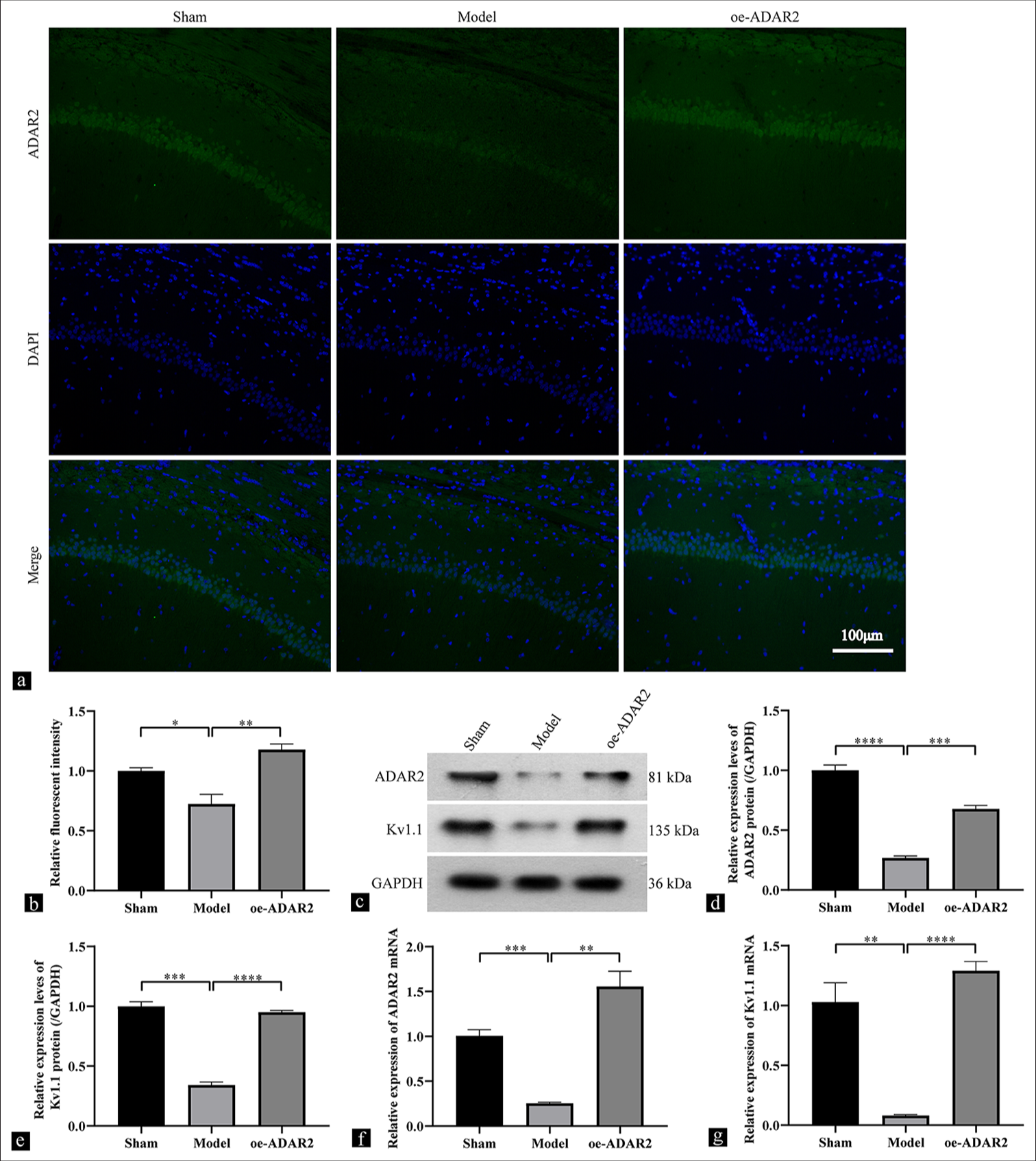
- Hippocampal ADAR2 and Kv1.1 expression changes. (a and b) Immunofluorescence results for ADAR2 in the sham, model, and oe-ADAR2 groups (n = 6). (c-e) Western blot and quantitative analyses of ADAR2 and Kv1.1 protein expressions (n = 6). (f and g) ADAR2 and Kv1.1 mRNA expressions detected via qRT-PCR (n = 3), magnification: ×200, scale: 100 mm. (**P < 0.01; ***P < 0.001, ****P < 0.0001. Sham: Sham, oe: Overexpression, ADAR2: Adenosine deaminase acting on RNA 2, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, Kv1.1: potassium voltage-gated channel subfamily A member 1, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, DAPI: 4¢,6-Diamidino-2-phenylindole).
Effect of ADAR2 overexpression on nerve damage in the hippocampus
Figure 2a illustrates HE-stained tissue sections of the hippocampal CA1 region. Light microscopy of the sham group revealed no evident impairment in hippocampal pyramidal cells. However, the model group exhibited an evident neuronal injury, which is characterized by cell loss, significant cell body shrinkage, and increased cell darkening. ADAR2 overexpression in the oe-ADAR2 group conferred a greater protection against neuronal death compared with the model group. Nissl staining revealed nuclear condensation or disaggregation and the loss of Nissl material in the hippocampus dentate gyrus of the model group. ADAR2 overexpression effectively mitigated cellular damage [Figure 2b]. NeuN-positive cells in the CA3 region of the hippocampus displayed similar alterations across all three groups [Figure 2c and d]. The model group exhibited a significantly lower percentage of positive neurons than the sham group (P < 0.001), and ADAR2 overexpression significantly reversed this trend (P < 0.001).
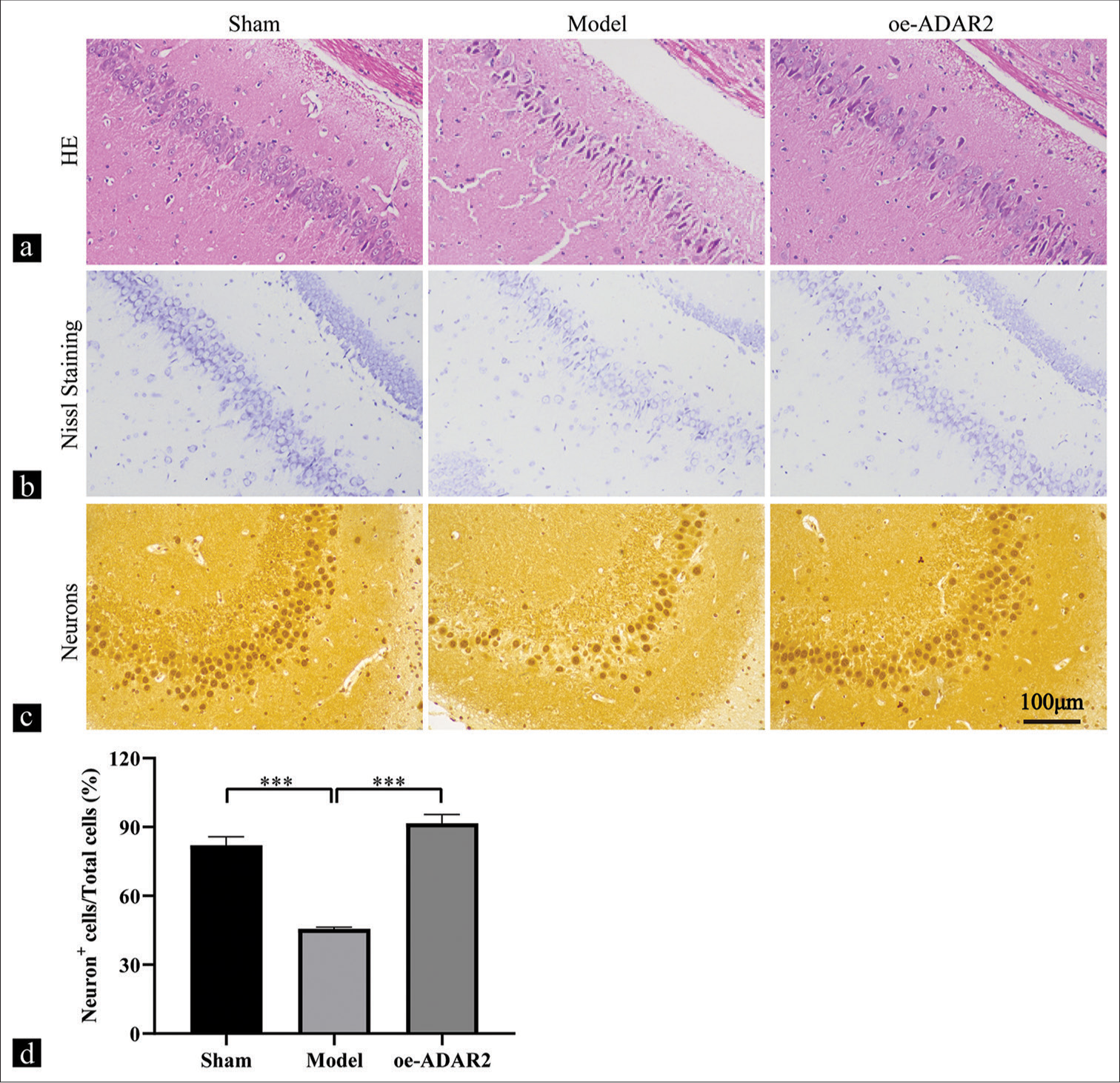
- Effects of ADAR2 overexpression on hippocampal neurons. (a) Hematoxylin and eosin (HE) staining, (b) Nissl staining; (c and d) immunohistochemical staining and quantitative analysis of neurons in the sham, model, and oe-ADAR2 groups (n = 6). Magnification: ×200; scale: 100 mm. ***P < 0.001. (Sham: Sham, oe: Overexpression, ADAR2: Adenosine deaminase acting on RNA 2, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, Kv1.1: Potassium voltage-gated channel subfamily A member 1, HE: Hematoxylin and eosin).
Effect of ADAR2 overexpression on the expression of apoptosis-related factors in the hippocampal subregion
Compared with that in the sham group, the model group showed a significantly increased in the Bax expression level, according Western blot analysis [Figure 3a-e] (P < 0.0001). This increase was reversed by ADAR2 overexpression (P < 0.0001). Conversely, the expression level of Bcl-2 exhibited an inverse pattern (P < 0.001). To investigate the potential involvement of ADAR2 overexpression in apoptosis, we evaluated the levels of cleaved caspases-3/7 in the hippocampal subregion. Western blot analysis revealed the significant upregulation of cleaved caspases-3/7 expression levels in the model group compared with the sham group (cleaved caspases-3, P < 0.001; cleaved caspases-7, P < 0.01). However, the levels of cleaved caspases-3/7 were decreased by ADAR2 overexpression (cleaved caspases-3, P < 0.0001; cleaved caspases-7, P < 0.01).
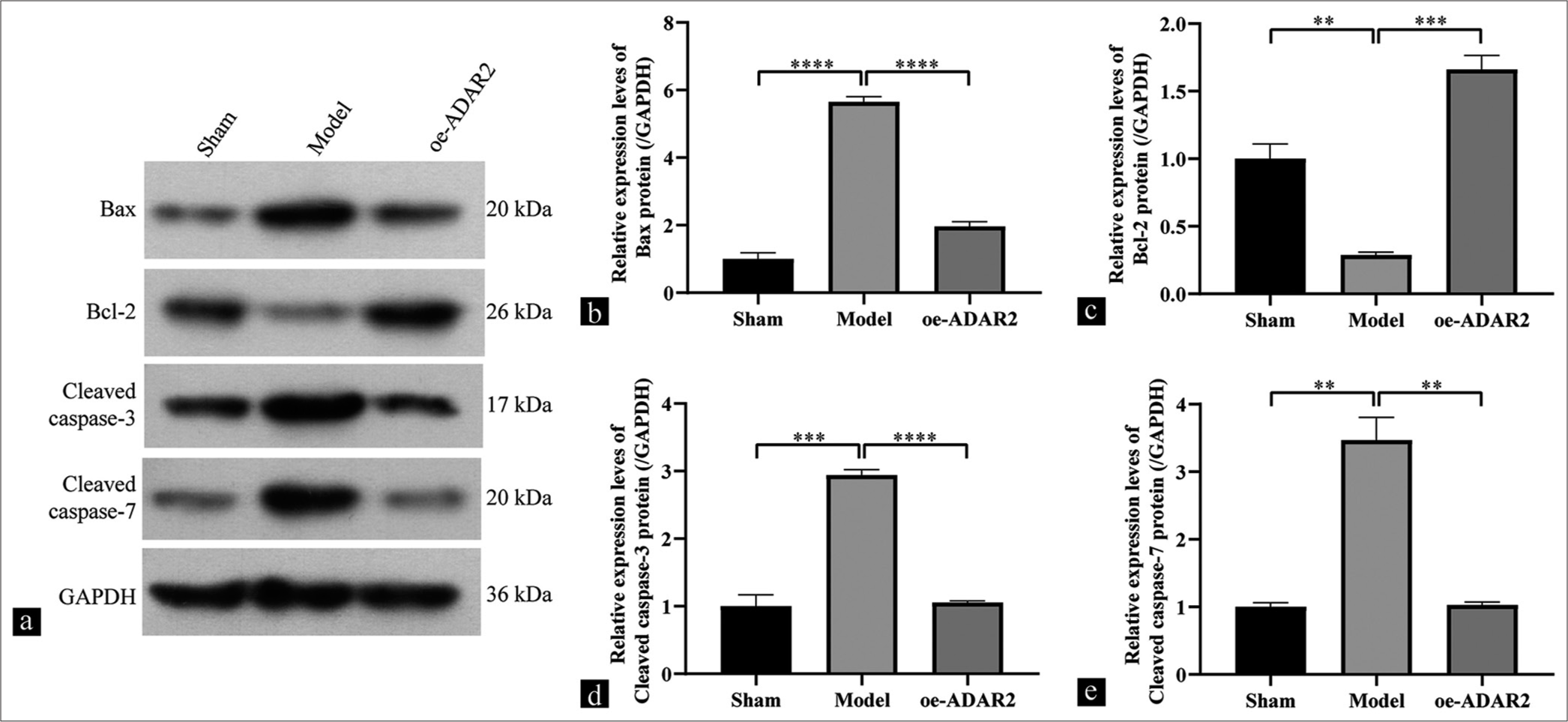
- Effect of ADAR2 overexpression on hippocampal Bax, Bcl-2, and cleaved caspases-3/7 expression. (a) Western blot bands of Bax, Bcl-2, and cleaved caspases-3/7. (b-e) Western blot analysis of Bax, Bcl-2, and cleaved caspases-3/7 (n = 3). **P < 0.01; ***P < 0.001, ****P < 0.0001. (Sham: Sham, oe: Overexpression, ADAR2: Adenosine deaminase acting on RNA 2, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, Kv1.1: Potassium voltage-gated channel subfamily A member 1, Bcl-2: B-cell leukemia/lymphoma 2 protein, Bax: Bcl-2-associated X protein).
Effect of ADAR2 overexpression on the expression of Kv1.1 and apoptosis-related factors in SH-SY5Y cells
SH-SY5Y cells were used to investigate the mechanism of ADAR2 regulation of Kv1.1. Western blotting analysis revealed that ADAR2, Kv1.1, and Bcl-2 expression levels exhibited a marked decrease in the EP group compared with the Con group (ADAR2, P < 0.01; Kv1.1, P < 0.05; Bcl-2, P < 0.001). However, ADAR2 overexpression significantly elevated the expression levels of Kv1.1 and Bcl-2 [Figure 4a-d, and e] (Kv1.1, P < 0.05; Bcl-2, P < 0.05). Western blotting analysis revealed the significantly higher expression levels of cleaved caspases-3/7 and Bax in the EP group compared with the Con group (Bax, P < 0.001; cleaved caspases-3, P < 0.01; Cleaved caspases-7, P < 0.01). This trend was reversed by ADAR2 overexpression [Figure 4d and f, g and h] (Bax, P < 0.001; cleaved caspases-3, P < 0.01; cleaved caspases-7, P < 0.01).
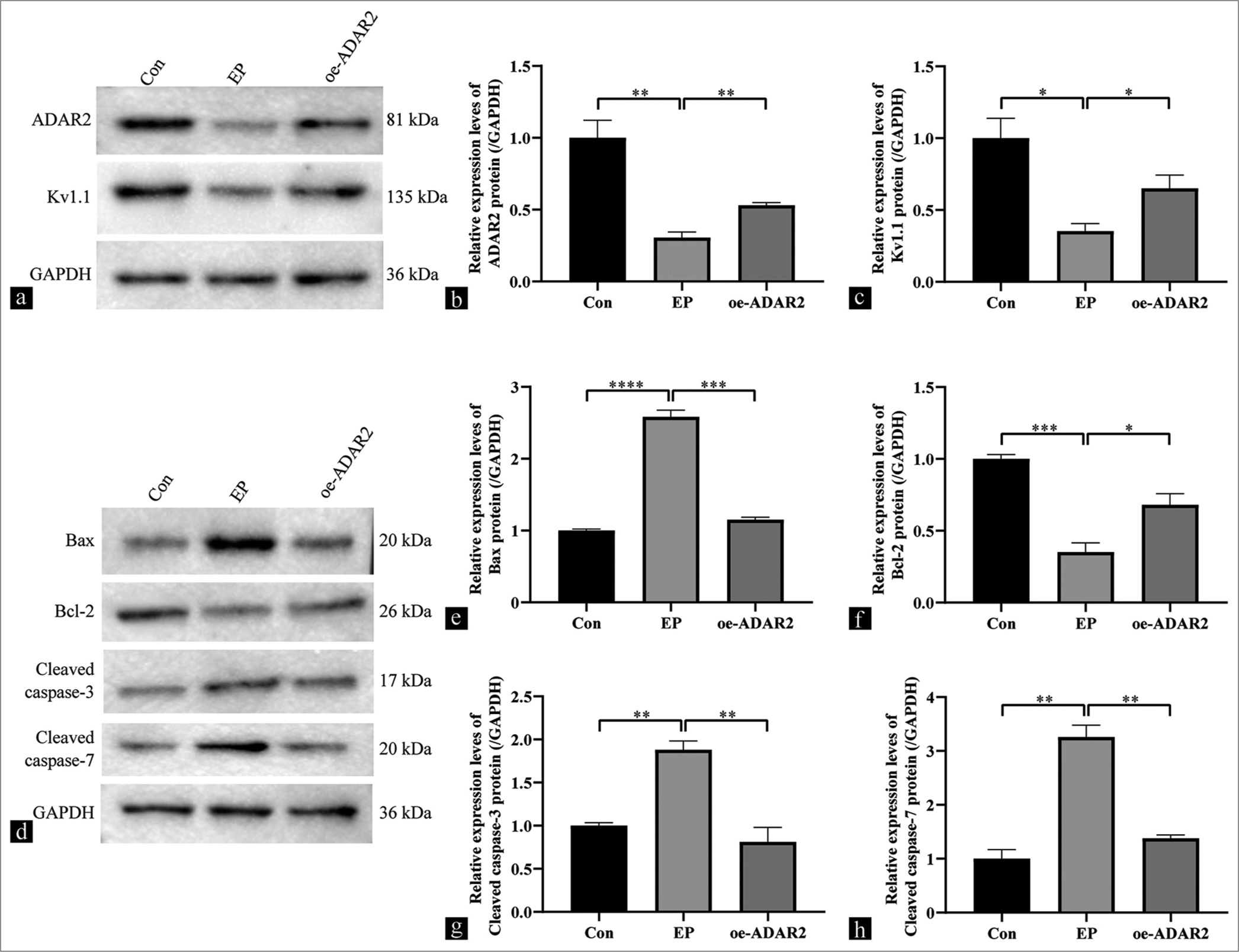
- Effects of ADAR2 overexpression on the expressions of Kv1.1, Bax, Bcl-2, and cleaved caspases-3/7 in SH-SY5Y cells from the Con, EP, and oe-ADAR2 groups. (a-c) Western blot bands and quantitative analysis of ADAR2 and Kv1.1. (d-h) Western blot bands and quantitative analysis of Bax, Bcl-2, and cleaved caspases-3/7 (n = 3). (*P < 0.05, **P < 0.01; ***P < 0.001, ****P < 0.0001. Con: Control, oe: Overexpression, EP: Epilepsy, ADAR2: Adenosine deaminase acting on RNA 2, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, Kv1.1: Potassium voltage-gated channel subfamily A member 1, Bcl-2: B-cell leukemia/lymphoma 2 protein, Bax: Bcl-2-associated X protein.).
To demonstrate the dependence of the regulatory function of ADAR2 in SH-SY5Y cells on the expression changes of Kv1.1, we knocked down Kv1.1 and transfected oe-ADAR2 cells to observe whether it can reverse the anti-apoptotic effect of ADAR2. First, the knockdown efficiency of Kv1.1 was verified through RT-PCR ([Figure 5a] (P < 0.0001). Subsequently, Western blotting was performed to examine the expressions of ADAR2 and Kv1.1. Compared with the oe-ADAR2 group, the oe-ADAR2 + si-Kv1.1 group showed no reduction in ADAR2 expression (ADAR2, ns, P > 0.05) but exhibited a decrease in Kv1.1 expression [Figure 5b-d] (Kv1.1, P < 0.01). Compared with the oe-ADAR2 group, the expression levels of cleaved caspases-3/7 and Bax were significantly increased in the oe-ADAR2 + si-Kv1.1 group (cleaved caspases-3, P < 0.05; cleaved caspases-7, P < 0.05), and that of Bcl-2 was significantly decreased ([Figure 5e-i] (P < 0.05). These findings indicate that ADAR2 regulated apoptosis in SH-SY5Y cells through the modulation of Kv1.1 expression.
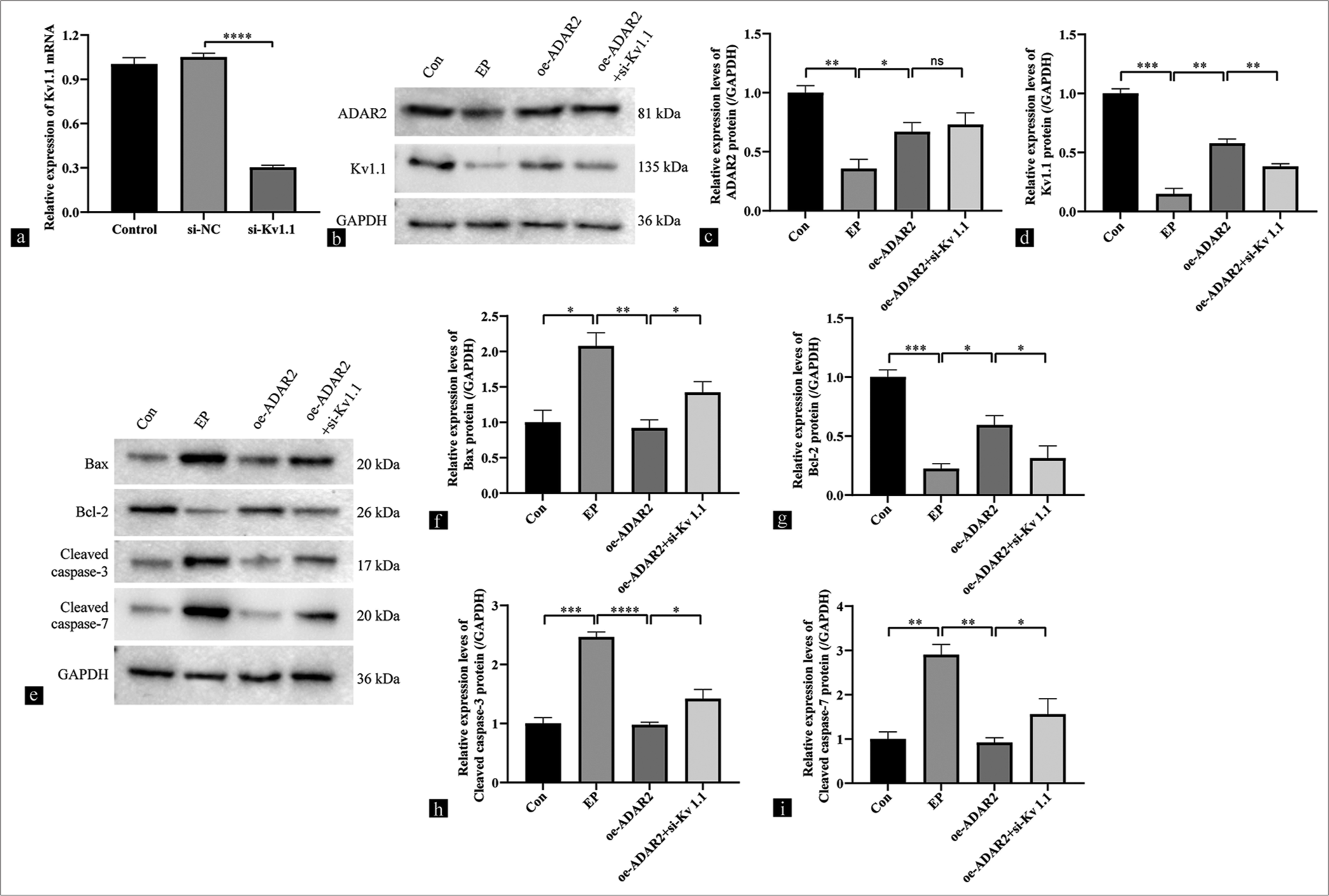
- Effects of ADAR2 overexpression on the expressions of Kv1.1, Bax, Bcl-2, and cleaved caspases 3 and 7 in SH-SY5Y cells from the Con, EP, oe-ADAR2, and oe-ADAR2+si-Kv1.1 groups. (a) Results on the knockdown efficiency of Kv1.1 by qRT-PCR. (b-d) Western blot bands and quantitative analysis of Kv1.1 and ADAR2. (e-i) Western blot bands and quantitative analysis of Bax, Bcl-2, and cleaved caspases-3/7 (n=3). (*P < 0.05, **P < 0.01; ***P < 0.001, ****P < 0.0001, ns: No significance. Si-NC: Si-negative control group, Con: Control, oe: Overexpression, EP: Epilepsy, ADAR2: Adenosine deaminase acting on RNA 2, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, Kv1.1: Potassium voltage-gated channel subfamily A member 1, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, Bcl-2: B-cell leukemia/lymphoma 2 protein, Bax: Bcl-2-associated X protein).
DISCUSSION
Epilepsy, which affects approximately 1.0% of the global population,[20] represents a complex neurological disease with a robust genetic component that involves numerous genes, with approximately a quarter of the genes encoding ion channels. Neurological conditions characterized by neuronal hyperexcitability, such as epilepsy, episodic ataxia type 1, and myokymia, show a possible association with the functionality of Kv1.1 voltage-gated potassium channels.[21] Our research supports the effectiveness of modulating the Kv1.1 channel to mitigate cellular damage in epilepsy models.
In Western blot and qRT-PCR analyses, significantly increased Kv1.1 protein and mRNA expression levels were observed in the hippocampus following the administration of oe-ADAR2 compared with the model group. Prior research linked hyperexcitability to the reduced Kv1.1 α-subunit expression, as observed in a mouse model of fragile X syndrome (FXS) featuring fragile X mental retardation gene 1 (Fmr1) deletion. Compared with age-matched wild-type mice, those with Fmr1 deletion presented a diminished Kv1.1 protein expression. Furthermore, patients with FXS exhibit heightened excitability in the central nervous system.
Wild-type neurons exhibit a notably increased average of evoked action potentials during the treatment with dendrotoxin-k (DTX-k), a Kv1.1-specific pharmacological blocker.[22] In addition, studies on apoptosis induction in rat hippocampal neurons with glutamate revealed a significant decrease in Kv1.1 mRNA and protein levels. Kv1.1 overexpression mitigated glutamate-induced apoptosis in the hippocampal neurons.[23] We further investigated the role of Kv1.1 in epilepsy by assessing its expression. This voltage-gated delayed potassium channel is encoded by the KCNA1 gene. Kv1.1 mRNA undergoes codon conversion mediated by ADAR, changing isoleucine to valine. ADAR2 predominantly mediates recoding editing in protein-coding genes, many of which are expressed in the central nervous system.[12] Figures 1b–1d illustrates the synchronous expression trends observed in Kv1.1 and ADAR2 protein and mRNA levels.
In this work, we did not delve into the specific splicing mechanism. To the best of our knowledge, a limited number of studies reported the effect of ADAR2 overexpression on nerve cells. We conducted in vivo and in vitro experiments to ascertain whether the RNA expression of ADAR2 is pivotal in regulating Kv1.1. Our findings indicate that ADAR2 influences Kv1.1 expression. The results of in vivo experiments suggest that ADAR2/Kv1.1 overexpression may improve neuronal survival by elevating Bcl-2 levels and reducing cleaved caspase-3/7 activity. However, in the recovery experimental group, silencing of Kv1.1 nullified the effect of ADAR2 overexpression.
The influence of ADAR2/kv1.1 on neuronal excitability and survival remains a controversy. The Kv1.1 gene regulation primarily affects the A-type potassium current in apoptotic neurons.[24] Apoptotic cerebellar granule neurons exhibit an elevated Kv1.1 protein expression.[25] According to literature, silencing of the Kv1.1 gene with small-interfering RNAs reduces the action potential amplitude but increases neuronal activity. In addition, the administration of DTX-κ significantly decreases Kv1.1 current amplitude and neuronal damage.[7] These findings imply a direct effect of Kv1.1 channel downregulation. Increased neuronal excitability may stem from ion-channel malfunction, which potentially causes clinical epileptic episodes. Substantial evidence confirms that variations in Kv1.1 expression exert undeniable electrophysiological effects that lead to cell damage and increase susceptibility to seizures.
Regional differences in Kv1.1 subunit complexes in the human central nervous system imply functional specialization.[26] These expression disparities may be attributed to the precise regulatory control of Kv1.1 across brain tissues or cell types. Alternatively, diverse functions of various Kv1.1 subunits in the brain can explain this variation following therapeutic interventions.
The Kv1.1 channel comprises alpha and beta subunits. According to earlier investigations, the voltage-dependent potassium channel formed by Kv1.1 and Kvb1.1 subunits produces a current characterized by fast-inactivating and sustained components. The dephosphorylation of phosphorylated Ser-446 on the alpha subunit can reduce the fast-inactivating component.[27] Moreover, Kv1.1 RNA expression can represent a critical determinant of cell survival or demise. These findings suggest that the Kv1.1 channel, which is regulated by a complex mechanism, influences nerve cell excitability and viability.
Current fundamental research on Kv1.1 channel-related illnesses often overlooks the role of adenosine-to-inosine (A-to-I) editing in Kv1.1. This editing, which is mediated by ADAR enzymes, fine tunes the genetic code and is contributes to the proper functioning of Kv1.1 channels, especially in the central nervous system. Although studies have shown variations in Kv1.1 editing across different brain regions, a comprehensive understanding of the mechanism underlying the effect of this editing on Kv1.1 function is lacking.
Human ADAR2 efficiently modifies a single adenosine in the Kv1.1 mRNA sequence.[28] Our animal experiments further confirmed that ADAR2 overexpression can elevate Kv1.1 expression level, which reduces nerve cell damage in the hippocampus of epileptic rats.
ADAR2 exhibits a prominently expression throughout the central nervous system, mainly within neuronal nuclei. This protein alters adenosines in pre-mRNAs upon nuclear entry. Binding to double-stranded RNA, ADAR2 deaminates specific A to I during Kv1.1 RNA processing. ADAR2 overexpression can upregulate Kv1.1 protein levels, which inhibits cell injury and reducing neuronal excitation.
Abnormal pharmacological and electrophysiological phenomena in epilepsy may partially originate from ADAR2 activation. The mechanism accounting for Kv1.1 RNA regulation by ADAR2 and the potential contributing factors remain subjects of inquiry. ADAR2-dependent glutamate receptor 2Q/R site editing serves as a key intracellular process that mitigates harmful signals during forebrain ischemia. In rat hippocampi, endogenous ADAR2 expression may offer protection against forebrain ischemic injury in susceptible neurons.[29] Concurrently, compromised Gamma-Aminobutyric Acid inhibitory signaling has been implicated in epilepsy and autism. In vertebrates, ADARs may perform an evolutionary conserved function that contributes to the regulation of neuronal excitability.[30] Our findings suggest that ADAR2 regulates neuronal survival through mediation of the expression of Kv1.1, which influences seizure onset and progression in epilepsy.
SUMMARY
Our findings confirmed the expression of ADAR2/Kv1.1 in neurons of hippocampus region. Our data also suggested that ADAR2 overexpression inhibits neuronal damage by promoting Kv1.1 protein expression in vivo and in vitro experiments.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
ABBREVIATION
Kv1.1 – Potassium voltage-gated channel subfamily A member 1
ADAR2 – Adenosine deaminase 2
qRT-PCR – Quantitative reverse transcription polymerase chain reaction
Bcl-2 – B-cell leukemia/lymphoma 2 protein
Bax – Bcl-2-associated X protein
HE – hematoxylin–eosin
CLSM – Confocal laser scanning microscope
DAPI – 4',6-Diamidino-2-phenylindole
PVDF – Polyvinylidene fluoride
GAPDH – Glyceraldehyde-3-phosphate dehydrogenase
AUTHOR CONTRIBUTIONS
PZ: Definition of intellectual content, experimental studies, data acquisition, data analysis, and manuscript preparation; WY: Concepts, literature search, experimental studies, data acquisition, data analysis, statistical analysis, manuscript preparation; WL: experimental studies, and data acquisition; JW: Experimental studies, statistical analysis; PW: Concepts, design, manuscript editing, and review; BN: Concepts, design, manuscript editing, and review. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All experimental procedures were approved by the Animal Ethics Committee of Guangzhou Red Cross Hospital (No. 2020-194-01; dated: September 1, 2020). Since this study does not involve human participants, consent to participate is not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This work was supported by the Guangdong Provincial Basic and Applied Basic Regional Joint Fund (2020A1515110009, PZ), Guangzhou Science and Technology Bureau City School (Institute) Enterprise Joint Project (2024A03J0653, PW), Guangzhou Science and Technology Bureau City School (Institute) Enterprise Joint Project (2024A03J0564, JW) and Research Grant of Key Laboratory of Regenerative Medicine, Ministry of Education, Jinan University (ZSYXM202305, PW).
References
- Potassium channels and epilepsy. Acta Neurol Scand. 2022;146:699-707.
- [CrossRef] [PubMed] [Google Scholar]
- Voltage-gated potassium channel blocker 4-aminopyridine induces glioma cell apoptosis by reducing expression of microRNA-10b-5p. Mol Biol Cell. 2018;29:1125-36.
- [CrossRef] [PubMed] [Google Scholar]
- Pharmacology of A-type K+ channels. Handb Exp Pharmacol. 2021;267:167-83.
- [CrossRef] [PubMed] [Google Scholar]
- Altered A-type potassium channel function in the nucleus tractus solitarii in acquired temporal lobe epilepsy. J Neurophysiol. 2019;121:177-87.
- [CrossRef] [PubMed] [Google Scholar]
- Upregulation of A-type potassium channels suppresses neuronal excitability in hypoxic neonatal mice. FEBS J. 2023;290:4092-106.
- [CrossRef] [PubMed] [Google Scholar]
- Blockade of voltage-gated potassium channels ameliorates diabetes-associated cognitive dysfunction in vivo and in vitro. Exp Neurol. 2019;10:112988.
- [CrossRef] [Google Scholar]
- Kv 1.1 is associated with neuronal apoptosis and modulated by protein kinase C in the rat cerebellar granule cell. J Neurochem. 2008;106:1125-37.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations underlying episodic ataxia type-1 antagonize Kv1.1 RNA editing. Sci Rep. 2017;7:41095.
- [CrossRef] [PubMed] [Google Scholar]
- Kv1.1 channelopathies: Pathophysiological mechanisms and therapeutic approaches. Int J Mol Sci. 2020;21:2935.
- [CrossRef] [PubMed] [Google Scholar]
- Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167-75.
- [CrossRef] [PubMed] [Google Scholar]
- ADAR family proteins: A structural review. Curr Issues Mol Biol. 2024;46:3919-45.
- [CrossRef] [PubMed] [Google Scholar]
- Adenosine-to-inosine RNA editing in neurological development and disease. RNA Biol. 2021;18:999-1013.
- [CrossRef] [PubMed] [Google Scholar]
- RNA editing in neurological and neurodegenerative disorders. Methods Mol Biol. 2021;2181:309-30.
- [CrossRef] [PubMed] [Google Scholar]
- Functional differences between two Kv1.1 RNA editing isoforms: A comparative study on neuronal overexpression in mouse prefrontal cortex. Mol Neurobiol. 2021;58:2046-60.
- [CrossRef] [PubMed] [Google Scholar]
- Adenosine-to-inosine RNA editing enzyme ADAR and microRNAs. Methods Mol Biol. 2021;2181:83-95.
- [CrossRef] [PubMed] [Google Scholar]
- RNA editing by ADAR adenosine deaminases: From molecular plasticity of neural proteins to the mechanisms of human cancer. Biochemistry (Mosc). 2019;84:896-904.
- [CrossRef] [PubMed] [Google Scholar]
- Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321-49.
- [CrossRef] [PubMed] [Google Scholar]
- Neural activities in multiple rat brain regions in lithium-pilocarpine-induced status epilepticus model. Front Mol Neurosci. 2020;12:323.
- [CrossRef] [PubMed] [Google Scholar]
- Intracerebroventricular and intravascular injection of viral particles and fluorescent microbeads into the neonatal brain. J Vis Exp. 2016;113:54164.
- [CrossRef] [Google Scholar]
- A review: Recent analytical applications on anti-epileptic agents. Curr Pharm Anal. 2022;9:825-40.
- [CrossRef] [Google Scholar]
- Case report: A novel loss-of-function pathogenic variant in the KCNA1 cytoplasmic N-terminus causing carbamazepine-responsive type 1 episodic ataxia. Front Neurol. 2022;13:975849.
- [CrossRef] [Google Scholar]
- Development-related aberrations in Kv1.1 a-subunit exert disruptive effects on bioelectrical activities of neurons in a mouse model of fragile X syndrome. Prog Neuropsychopharmacol Biol Psychiatry. 2018;84:140-51.
- [CrossRef] [Google Scholar]
- Contribution of Kv channel subunits to glutamate-induced apoptosis in cultured rat hippocampal neurons. J Neurosci Res. 2009;87:3153-60.
- [CrossRef] [PubMed] [Google Scholar]
- Kv1.1 null mice have enlarged hippocampus and ventral cortex. BMC Neurosci. 2007;8:10.
- [CrossRef] [PubMed] [Google Scholar]
- Loss of functional system x-c uncouples aberrant postnatal neurogenesis from epileptogenesis in the hippocampus of Kcna1-KO mice. Cell Rep. 2022;41:111696.
- [CrossRef] [PubMed] [Google Scholar]
- Kv1 potassium channels control action potential firing of putative GABAergic deep cerebellar nuclear neurons. Sci Rep. 2020;10:6954.
- [CrossRef] [PubMed] [Google Scholar]
- Kv1.1 channel subunits in the control of neurocardiac function. Channels (Austin). 2019;13:299-307.
- [CrossRef] [PubMed] [Google Scholar]
- RNA editing of a human potassium channel modifies its inactivation. Nat Struct Mol Biol. 2004;11:915-6.
- [CrossRef] [PubMed] [Google Scholar]
- ADAR2-Dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49:719-33.
- [CrossRef] [PubMed] [Google Scholar]
- The ADAR RNA editing enzyme controls neuronal excitability in Drosophila melanogaster. Nucleic Acids Res. 2014;42:1139-51.
- [CrossRef] [PubMed] [Google Scholar]




