


Translate this page into:
CytoJounal quiz case: Fine-needle aspiration of peripancreatic mass clinically mimicking a lymphoma
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
A 74-year-old male is found to have hypermetabolic upper abdominal lymphadenopathy clinically suspicious for lymphoma. After obtaining the consent, fine-needle aspiration (FNA) of the mass is performed, and ThinPrep slide and a cell block are prepared for evaluation. ThinPrep shows atypical cells with abundant pale cytoplasm, stippled chromatin, and mild anisonucleosis. The cell block shows clusters of cells with pale cytoplasm and somewhat granulomatous appearance [Figure 1]. An exhaustive immunohistochemical panel shows cells negative for AE1/3, TTF1, Hep Par, CD21, Cd1a, DOG1, inhibin, S100, and calretinin. The cells are positive for synaptophysin (image included) and chromogranin [Figure 2]. Rare cells are positive for SOX10.
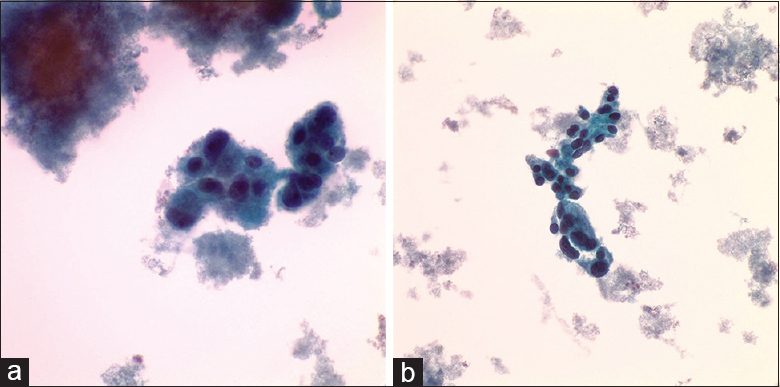
- (a) ThinPrep (×400) shows cells with abundant cytoplasm and stippled chromatin. (b) ThinPrep (×400) – the cells exhibit mild anisonucleosis
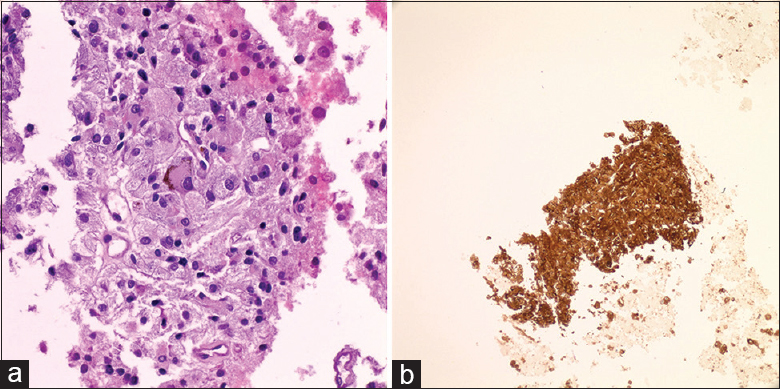
- (a) Cell block reveals cluster of cells with pale cytoplasm and granuloma-like formation. (b) Immunohistochemistry for synaptophysin
QUESTION
What is your interpretation?
-
Metastatic neuroendocrine carcinoma
-
Solid pseudopapillary carcinoma
-
Adrenal cortical carcinoma
-
Paraganglioma
-
Melanoma.
ANSWER AND FOLLOW-UP OF THE CASE
Based on the cytologic features, immunohistochemistry results, and location, the correct interpretation is C. paraganglioma.
The differential diagnosis based on the careful morphologic examination and location of the tumor includes hepatocellular carcinoma, pancreatic neuroendocrine tumor, adrenocortical carcinoma, melanoma, epithelioid gastrointestinal tumor, histiocytic lesions, and paraganglioma.
Paraganglioma is a rare tumor arising from the chromaffin cells in extra-adrenal sites. They can arise in all age groups with a peak incidence in the 4th and 5th decades. Cytologic diagnosis of extra-adrenal paraganglioma presenting as a peripancreatic mass is challenging with a high error rate due to its rarity.[1] Diagnosis on cytologic (FNA specimens) is usually challenging. To render the correct diagnosis, the pathologist should keep in mind the anatomic location and cytologic features reminiscent of an endocrine neoplasm. If the possibility of paraganglioma arises during on-site evaluation, it is necessary to collect cell block material and perform the immunostains to establish the diagnosis.[2] The diagnosis can be correctly established when the lesion is morphologically suspected on smears/Thinprep and cell block material. The tumor is positive for neuroendocrine markers, and usually, S100 protein is immunoreactive in sustentacular cells. However, in this case, S100 is noncontributory, but SOX10 is positive in rare sustentacular cells. In addition, succinate dehydrogenase (SDH) beta subunit is retained by immunohistochemistry in our case.
ADDITIONAL QUIZ QUESTIONS
Q2. How would you classify tumor of adrenal medulla arising from the chromaffin cells and how often are they malignant?
-
Paraganglioma: 30% are malignant
-
Pheochromocytoma: 90% are malignant
-
Pheochromocytoma: 10% are malignant
-
Cortical adrenal tumor: 40% are malignant.
Q3. In addition to immunohistochemical findings for diagnostic purposes, what other immunohistochemistry stain is recommended if available?
-
Catecholamine
-
Cytokeratin
-
Melan A
-
SDH beta subunit.
Q4. Paraganglioma syndrome is associated with following syndromes:
-
von Hippel–Lindau (VHL)
-
Carney triad
-
Alport syndrome
-
Pheochromocytoma/paraganglioma syndrome
-
A, B, D
-
All of the above.
ANSWERS AND BRIEF REVIEW OF THE TOPIC
Q2: C Q3: D Q4: E
Neoplasms arising from the chromaffin cells in the adrenal medulla are classified as pheochromocytoma. Both paraganglioma and pheochromocytoma can produce and secrete catecholamines, thus presenting with hypertension.[3] About 90% of pheochromocytoma are benign and about 10% are bilateral. In typical paraganglioma/pheochromocytoma presentations (with hypertension), the first line of diagnosis is computed tomography (CT) scan. Functional imaging with 123I-MIBG or 18F-fludeoxyglucose positron emission tomography/CT is also acceptable if no lesion can be identified by morphological cross-sectional imaging or if metastatic disease is suspected.
Octreoscan is excellent in the investigation of paraganglioma and neuroendocrine tumors in general.[4]
In benign lesions, surgery is the first line of treatment.
Paragangliomas can be sporadic or familial and associated with various syndromes such as VHL disease, Carney triad, neurofibromatosis type 1, MEN 2A and 2B, and hereditary pheochromocytoma/paraganglioma syndrome associated with SDH gene mutations. Familial forms of pheochromocytoma/paraganglioma syndrome can be frequently clinically recognized by the younger age of onset (<45), multiple and multifocal tumors, recurrent tumors, and family history of such tumors.
It is currently thought, according to some studies, that 65%–80% of all pheochromocytomas/paragangliomas are associated with somatic or germline mutations and some can further be associated with one of the above syndromes.[5] The American Society of Clinical Oncology recommends offering genetic screening for all patients with a risk of at least 10% of carrying a genetic mutation, especially when the results aid in diagnosis or influence the management of the patient or family members at hereditary risk of cancer.[6] Some authors suggest that genetic screening should be performed in all patients with a paraganglioma.[5]
Pheochromocytoma/paraganglioma syndrome is associated with a mutation in one SDH subunit that plays a critical role in mitochondria.
Immunohistochemistry for SDHB has been shown to be an excellent screening tool for a mutation in SDH genes. Immunohistochemistry for SDHB is lost whenever there is a complete inactivation of SDHA, SDHB, SDHC, SDHD, or SDHAF2. As a result, loss of tumoral immunohistochemical staining for SDHB occurs when there is germline mutation of SDHA, SDHB, SDHC, or SDHD accompanied by inactivation of the normal allele. This makes loss of staining for SDHB a sensitive marker, suggestive of germline pathogenic variants of any of the SDH subunits.[78] Germline pathogenic variants in SDHA show loss of staining for SDHA, in addition to loss of staining for SDHB.[9] In conclusion, negative/loss of staining for SDHB is an absolute indication for genetic testing and also indicates high risk of malignant behavior.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
COMPETING INTERESTS STATEMENT BY ALL AUTHORS
The authors declare that they have no competing interests.
AUTHORSHIP STATEMENT BY ALL AUTHORS
Adela Cimic MD-primary author, data collection and drafting of the manuscript. Natasha Rekhtman, MD, senior author, data review and review of the manuscript.
ETHICS STATEMENT BY ALL AUTHORS
This study was conducted with approval from the Institutional Review Board (or its equivalent) of all the institutions associated with this study as applicable. Authors take responsibility to maintain relevant documentation in this respect.
LIST OF ABBREVIATIONS (In alphabetic order)
CT - Computed tomography,
FNA - Fine needle aspiration,
SDH - Succinate dehydrogenase,
VHL - Von Hippel-Lindau.
EDITORIAL/PEER-REVIEW STATEMENT
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (the authors are blinded for reviewers and vice versa) through automatic online system.
REFERENCES
- Paraganglioma: Cytomorphologic features, radiologic and clinical findings in 12 cases. Diagn Cytopathol. 2018;46:473-81.
- [Google Scholar]
- Diagnostic challenges in the fine-needle aspiration diagnosis of carotid body paragangliomas: Report of two cases. Diagn Cytopathol. 2000;23:202-7.
- [Google Scholar]
- Paraganglioma or pheochromocytoma? A peculiar diagnosis. J Surg Case Rep. 2018;4:1-4.
- [Google Scholar]
- Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: A multicenter prospective study from the PGL. EVA investigators. J Clin Endocrinol Metab. 2013;98:162-73.
- [Google Scholar]
- Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015;52:647-56.
- [Google Scholar]
- Paragangliomas/pheochromocytomas: Clinically oriented genetic testing. Int J Endocrinol. 2014;2014:794187.
- [Google Scholar]
- Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41:805-14.
- [Google Scholar]
- Usefulness of succinate dehydrogenase B (SDHB) immunohistochemistry in guiding mutational screening among patients with pheochromocytoma-paraganglioma syndromes. APMIS. 2014;1122:1130-5.
- [Google Scholar]
- Genetic analyses of apparently sporadic pheochromocytomas: The rotterdam experience. Ann N Y Acad Sci. 2006;1073:138-48.
- [Google Scholar]




