


Translate this page into:
Enhanced efficacy of dual chimeric antigen receptor-T cells targeting programmed death-ligand 1 and cancer-associated fibroblasts in colorectal cancer in vitro
 PhD, QiaoJin Xie1MM, HaoJie He1BD, JiaWei Li3MM, JiDong Miao3MM
PhD, QiaoJin Xie1MM, HaoJie He1BD, JiaWei Li3MM, JiDong Miao3MM

*Corresponding author: Yang Gao, Health Management Center, Zigong Fourth People’s Hospital, Zigong, Sichuan Province, China. 18882030231@163.com
-
Received: ,
Accepted: ,
How to cite this article: Gao Y, Luo C, Yang H, Xie Q, He H, Li J, Miao J, et al. Enhanced efficacy of dual chimeric antigen receptor-T cells targeting programmed death-ligand 1 and cancer-associated fibroblasts in colorectal cancer in vitro. CytoJournal. 2025;22:29. doi: 10.25259/Cytojournal_245_2024
Abstract
Objective
Colorectal cancer (CRC) presents significant treatment challenges, including immune evasion and tumor microenvironment (TME) suppression. Chimeric antigen receptor (CAR) T-cell therapy has shown promise in hematologic malignancies, but its effectiveness against solid tumors is hampered by the detrimental effects of the TME. This article aims to explore the potential of bispecific CAR T cells targeting programmed death-ligand 1 (PD-L1) and cancer-associated fibroblasts (CAFs) in CRC treatment.
Material and Methods
Dual-targeted CAR-T cells against PD-L1 and CAF were engineered using the GV400 lentiviral vector. Programmed death-1 (PD-1)/nanobody (Nb) and fibroblast activation protein (FAP)/Nb-encoding lentiviral vectors were generated, and CAR T cells were produced through a three-plasmid system in 293T cells. Human peripheral blood mononuclear cells (PBMCs) were separated, transduced with these vectors, and then expanded. Functional characterization of CAR-T cells was performed through enzyme-linked immunosorbent assay (ELISA), Western blot analysis, flow cytometry, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, and cell counting kit-8 (CCK-8) assay. Migration and invasion assays were conducted using Transwell chambers to assess the ability of FAP-PD-1/Nb CAR-T cells to migrate toward tumor cells and invade the extracellular matrix.
Results
We developed dual-targeted CAR-T cells incorporating PD-L1 and CAF Nbs, which continuously secreted PD-1/Nb. Western blot confirmed PD-1/Nb expression in PD-1/Nb and FAP-PD-1/Nb CAR-T cells, with no expression in the untreated (UTD) group (P < 0.01). Flow cytometry showed a significantly higher cluster of differentiation (CD)25 and CD69 expression in FAP-PD-1/Nb CAR-T cells upon stimulation with FAP-positive target cells compared with the other groups (P < 0.01). TUNEL, flow cytometry, and CCK-8 assays revealed that FAP-PD-1/Nb CAR-T cells exhibited superior cytotoxicity and proliferation inhibition against FAP-positive HCT116 cells (P < 0.01). ELISA demonstrated increased interferon-gamma and tumor necrosis factor-alpha levels and reduced interleukin-10 (P < 0.01), suggesting enhanced cytokine modulation and antitumor immunity. Compared with single-target CAR-T cells and UTD, FAP-PD-1/Nb CAR-T cells showed notably enhanced Matrigel penetration and invasion (P < 0.01). Safety tests confirmed minimal cytotoxicity to normal PBMCs, indicating favorable safety.
Conclusion
This study successfully developed dual-targeted CAR-T cells against PD-L1 and CAF and demonstrated their superior antitumor activity and immunomodulatory effects on CRC treatment. This novel therapeutic strategy was established using CAR T-cell technology for the treatment of CRC.
Keywords
Chimeric antigen receptor-T cells
Colorectal cancer
Fibroblast activation protein
Programmed death-ligand 1
Tumor microenvironment

INTRODUCTION
Colorectal cancer (CRC) ranks as the third most prevalent malignancy globally and comprises 10% of new cancer diagnoses; it is a major cause of cancer mortality and accounts for 9.4% of all cancer-related deaths.[1] Despite significant advancements in CRC treatment modalities such as surgery, chemotherapy, radiotherapy, and targeted therapies, these conventional approaches still face considerable challenges. The complex and immunosuppressive tumor microenvironment (TME), the presence of multiple immune checkpoint molecules, and the limited repertoire of highly specific tumor antigens hinder treatment efficacy.[2] Advanced CRC has a dismal prognosis, demonstrating a 5-year survival rate below 10% and limited overall survival improvements.[3] As such, novel therapeutic approaches are urgently needed.
The TME in CRC is a heterogeneous ecosystem that includes tumor cells, immune cells, endothelial cells, fibroblasts, and components of the extracellular matrix.[4] A major feature of the CRC TME is its high degree of immune suppression, which is largely driven by tumor cells expressing immune checkpoint molecules, such as programmed death-ligand 1 (PD-L1). These molecules not only inhibit immune cell function but also promote tumor progression and metastasis.[5] Furthermore, CRC tumors are characterized by an abundance of cancer-associated fibroblasts (CAFs), especially those expressing fibroblast activation protein (FAP). CAFs contribute to tumor progression by secreting extracellular matrix components that create physical barriers to immune cell infiltration and enhance tumor cell invasion and metastasis.[6] This immunosuppressive and barrier-rich microenvironment significantly complicates the treatment of CRC.
Chimeric antigen receptor (CAR) T-cell immunotherapy has recently achieved significant success against hematologic malignancies. Genetically engineered T cells expressing CARs selectively target and eradicate cancer cells through major histocompatibility complex (MHC)-independent recognition of tumor-associated antigens.[7] However, the treatment of solid tumors, such as CRC with CAR T-cell therapy, presents unique challenges. These include the heterogeneous antigen expression profiles of solid tumors and the immune-suppressive TME, both of which contribute to reduced CAR-T cell efficacy and potential toxicity.[8] Efforts to modify CAR-T potency in malignancies have focused on modifying CAR constructs, including dual-target or tandem CAR strategies, to simultaneously target multiple tumor antigens.[9]
The programmed death-1 (PD-1)/PD-L1 axis is a key immunosuppressive mechanism in CRC, and high PD-L1 expression in CRC cells correlates with tumor progression, metastasis, and poor prognosis.[10] Targeting this axis offers an effective approach to restore antitumor immunity, with several PD-1/PD-L1 inhibitors showing clinical efficacy in CRC.[11] In addition, CAFs in the CRC TME, characterized by high levels of FAP expression, significantly contribute to immune evasion and progression of CRC. Targeting FAP suppresses CAF activity and enhances immune responses against tumors in preclinical studies.[12] The present study investigates dual-targeted CAR T cells against PD-L1 and CAFs to enhance antitumor efficacy in CRC. The strategy aims to overcome key challenges in CRC immunotherapy by simultaneously blocking the PD-1/PD-L1 checkpoint and inhibiting CAF function. In vitro evaluation of these dual-targeted CAR T cells will determine their cytotoxic effects on CRC cell lines and explore their ability to modulate immune responses. Results will provide a basis for advancing dual-targeted CAR T-cell therapies as a promising treatment for advanced CRC.
MATERIAL AND METHODS
Construction of CAR-T cells
Construction of lentiviral vectors
CAR-T cells, including a His-tag for protein purification were produced using the GV400 lentiviral vector system, as described by Yang.[13] The GV400 vector was digested with BamHI/EcoRI enzymes. A polymerase chain reaction (PCR)-amplified gene fragment containing the FAP-PD-1/Nanobody (Nb) CAR structure (FAP-PD-1/Nb CAR-F: AGGTCGACTCTAGAGGATCCCGCCACC; FAP-PD-1/Nb CAR-R: ATAAGCTTGATATCGAATTCTTACTTGTACA GCTCGTCCATGCCGAG) was obtained. This gene fragment was then ligated into the digested vector and transformed into competent Escherichia coli cells. After colony PCR identification and positive clone sequencing, successful insertion of the target gene into the gene vector 400 was confirmed. The sequences of the vectors were verified by Sanger sequencing to ensure accuracy. The vectors for PD-1/Nb CAR and FAP/Nb CAR were synthesized by Sangon (Beijing, China).
PD-1/Nb CAR -F: AGGTCGACTCTAGAGGAT CCCGCCACCATGGCCTTACCAGTGACC GCCTTG;
PD-1/Nb CAR -R: GCGCCGCTGCCCATGGATCCAT GATGATGATGATGATGTGAGGAG;
FAP/Nb -CAR F: ACTAGTATGGAGAGCCCGC TGACC;
FAP/Nb -CAR R: GGGCCCTTATTTATCATCATCTT TATAATCTTAGCGCGGC.
Lentivirus was packaged using a three-plasmid system (envelope plasmid, packaging plasmid, and vector plasmid) in 293T cells (CL-0005, Pricella, Wuhan, China). At 24 h before transfection, logarithmic-phase 293T cells were digested with trypsin in a transfection kit (10668-006, Biomedicine, Chongqing, China), collected, and reseeded into culture dishes. At 2 h before transfection, 293T cells were cultured in serum-free Dulbecco’s modified Eagle medium (C11995500BT, ThermoFisher Scientific, Waltham, MA, USA). The three plasmids were mixed with Lipofectamine 2000 (11668500, ThermoFisher Scientific, Waltham, MA, USA) and added to the 293T cell culture. After incubation for 2-3 days (37°C, 5% carbon dioxide [CO2]), the transfected-cell supernatant was harvested, filtered (0.22 µm), and concentrated. The resulting lentiviral particles were resuspended in sterile solution and cryopreserved at −80°C.
T cell culture and transfection
Human peripheral blood mononuclear cells (PBMCs) (50 mL, Primary Cell Solutions-800-011, American Type Culture Collection) were resuspended in Roswell Park Memorial Institute (RPMI) medium 1640 (L211013, Basalmedia Biotechnology, Shanghai, China) (10% fetal bovine serum) (AC03L055, Life-iLab Biotechnology, Shanghai, China) and incubated (37°C, CO2). After centrifugation, T cells were cultured with interleukin (IL)-2 for 24 h. Lentivirus-containing supernatant was added, and green fluorescent protein fluoresce (GFP) fluorescence was monitored at 48, 72, and 96 h to assess infection efficiency. Mycoplasma detection and short tandem repeat (STR) analysis were performed on the cell lines used. The results indicated that the cells were not contaminated with mycoplasma, and STR analysis confirmed the identity of the cells.
Enzyme-linked immunosorbent assay (ELISA) for PD-1/Nb expression in CAR-T cell supernatants
PD-1/Nb supernatant levels were quantified using a one-step sandwich ELISA at multiple time points (days 1-14). The supernatants from different time points were collected and stored at −20°C for future analysis. Before use, all reagents were equilibrated to room temperature. The 96-well microplate was coated with protein (His-tagged PD-1/Nb or standards) and incubated overnight at 4°C. After incubation, the plate was washed 5 times with washing buffer, each wash lasting 1 min. Diluted samples and standards of various concentrations were then added to the corresponding wells, with 100 µL per well. The plate was sealed and incubated at 25°C for 20–30 min. Following incubation, the liquid was discarded, and the plate was washed again 5 times. Each well was then added with 100 µL of diluted HisTag monoclonal antibody (MAB12807, Biomart, China), and the plate was incubated at 25°C for 20–30 min. After washing, 100 µL of secondary anti-mouse immunoglobulin G (IgG)- horseradish peroxidase antibody (HRP) antibody (cluster of differentiation [CD]-1657Y, Solarbio, China) was added, and the plate was incubated at 25°C for another 20–30 min. The plate was washed 5 times, and 100 µL of 3,3’,5,5’-tetramethylbenzidine substrate (TMB) substrate (PR1210, Solarbio, Beijing, China) was added to each well, followed by incubation at room temperature for 5–10 min. The reaction was stopped by adding 100 µL of stop solution, and absorbance at 450 nm was recorded using a microplate reader (CMax Plus, Molecular Devices Instruments Co., Ltd., USA). PD-1/Nb concentrations in the samples were determined from a standard curve constructed from the absorbance values of the standards.
Western blot for PD-1/Nb expression in CAR-T cells
Total protein was extracted from PD-1/Nb, FAP/Nb, FAP-PD-1/Nb, and untreated (UTD) CAR-T cells using radioimmunoprecipitation assay buffer (R0010, Solarbio, Beijing, China) and clarified by centrifugation (13000 rpm, 20 min). Protein concentration was determined using the bicinchoninic acid protein (BCA) assay kit (P0010, Beyotime, Shanghai, China) according to the manufacturer’s instructions. The samples were clarified by centrifugation (13000 rpm, 20 min) and prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by mixing 500 µg of total protein with ×5 loading buffer. After electrophoresis, the proteins were transferred onto a polyvinylidene difluoride membrane (1704156, Bio-Rad, Hercules, CA, USA). The transfer was carried out using the Trans-Blot Turbo Transfer System (1704150, BioRad, Hercules, CA, USA) according to the manufacturer’s instructions. The membrane was blocked with 5% nonfat milk in tris-buffered saline with tween 20 (TBST) (PS103, Epizyme Biology, Shanghai, China) for 1 h at room temperature. The membrane was incubated overnight at 4°C with His-tag antibody (1:1000, MAB12807, Biomart) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (A19056, ABclonal Technology, Wuhan, China), which specifically binds to the His-tag fusion protein present on PD-1/Nb, to detect PD-1/Nb expression. A secondary HRP-labeled goat anti-rabbit IgG (1:2000, SE134, Solarbio, Beijing, China) was used to detect the His-tag signal, and the plate was incubated at 25°C for another 30 min. After washing with TBST, the membrane was treated with an enhanced chemiluminescence substrate (34580, Thermo, Waltham, MA, USA) and visualized using a gel imaging system (Universal Hood II, Bio-Rad, USA). Band density was quantified using ImageJ software (1.48 b, National Institutes of Health, Bethesda, MD, USA) for relative expression analysis.
Culture of CRC and normal cell lines
PBMCs were resuspended in phosphate-buffered saline (PBS) (G0002, Servicebio Biotechnology, Wuhan, China) and centrifuged again. PBMCs (105 cells/mL) were seeded in six-well plates and co-cultured with FAP-PD-1/Nb CAR-T cells (RPMI 1640, 37°C, 5% CO2) for 72 h. The media were refreshed as required, and control groups were set. HCT116 (CL-0096, Procell, China) and SW480 (CL-0223, Procell, China) cells were co-cultured with CAR-T cells (1:1 ratio). These cell lines were selected because they are relevant to CRC and are commonly used models for studying CRC biology. Both HCT116 and SW480 cell lines exhibit strong expression of FAP, a marker associated with CAFs in TME.[14] We performed mycoplasma detection and STR analysis on the cell lines used. The results indicated that the cells were not contaminated with mycoplasma, and STR analysis confirmed the identity of the cells.
Flow cytometry
FAP-PD-1/Nb CAR-T cells were prepared and adjusted to a density of 1 × 106 cells/mL. The CAR-T cells were co-cultured with target cells (HCT116 and SW480) for stimulation. After co-culturing, target cells were collected and digested with ethylenediaminetetraacetic acid (EDTA)-free trypsin (without phenol red) (15090046; Gibco, USA) and resuspended in culture medium. The cells were then centrifuged at 1000 rpm for 5 min, and the supernatant was discarded. The cells were washed with PBS and centrifuged again. The supernatant was discarded, and the residue was washed 3 times. For flow cytometry analysis of FAP expression, the cells were incubated with 5 µL of FAPfluorescein isothiocyanate (FITC) flow cytometry antibody and 50 µL of antibody binding buffer for 30 min in the dark. After incubation, the cells were washed 3 times with PBS and resuspended at the appropriate cell density. FAP expression was analyzed by flow cytometry, and a negative control group was included for comparison. Following the co-culture of CAR-T cells with FAP-positive target cells, T lymphocytes were collected. The cells were stained with 0.5 µg of phycoerythrin (PE)-conjugated anti-CD25/APC (302605, BioLegend, China) and incubated at 4°C for 30 min. After washing twice with 2 mL of flow cytometry wash buffer, the cells were centrifuged at 1000 rpm for 5 min, and the supernatant was discarded. The cells were then resuspended in 100 µL of wash buffer, added with 0.5 µg of PEconjugated anti-CD69/FITC (310905, BioLegend, China), and incubated for 30 min at 4°C. A PE-conjugated isotype control antibody (400111, BioLegend, China) was used for the control group. After incubation, the cells were washed twice with 2 mL of wash buffer, centrifuged at 1000 rpm for 5 min, and the supernatant discarded. Finally, the cells were resuspended in 500 µL of wash buffer, and flow cytometric analysis (CytoFLEX, Beckman Coulter, Brea, CA, USA) was performed.
For apoptosis detection, 1 × 106 cells were centrifuged again at 1000 rpm for 3 min, and the supernatant was removed. The cells were then incubated with 200 µL of annexin V-FITC-A binding solution (40302ES20, Yeasen Biotechnology, Shanghai, China) and gently resuspended. After 10 min of incubation at 25°C in the dark, the cells were centrifuged at 1000 rpm for 3 min, and the supernatant was discarded. The cells were then treated with an additional 190 µL of annexin V-FITC-A binding solution and 10 µL of propidium iodide staining solution (C1056, Beyotime, Shanghai, China) and kept on ice in the dark. Flow cytometry analysis was performed using a CytoFLEX system (CytoFLEX, Beckman Coulter, Brea, CA, USA) to assess cell apoptosis.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay for apoptosis
Cell paraffin sections were prepared and fixed with 4% paraformaldehyde. The fixed cells were permeabilized (0.3% Triton X-100: ST795, Beyotime, Shanghai, China, 5 min), stained using a TUNEL apoptosis detection kit (CY3 red fluorescence: G1502, Servicebio Biotechnology, Wuhan, China), and washed. The nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (C1005, Beyotime Biotechnology, Shanghai, China), and an anti-fade reagent was applied. Apoptotic cells were detected under a fluorescence microscope (ICX41, Sunny Optical Technology [Group] Co Ltd., China), with positive apoptotic nuclei showing red fluorescence and DAPI staining nuclei blue.
Cell counting kit-8 (CCK-8) assay
Cells were seeded in 96-well plates (12 h incubation) and treated with CCK-8 solution (10 µL/well, 4 h incubation) (C0038, Beyotime, Shanghai, China). Absorbance (450 nm) was recorded using a microplate reader (CMax Plus, Molecular Devices Instruments Co., Ltd., USA). Cell viability (%) was calculated as ([optical density (OD)experimental –
ODblank]/[ODcontrol – ODblank]) × 100%, where experimental, blank, and control groups represent wells containing cells with overexpressed/interfered plasmids, media alone, and empty-vector controls, respectively.
ELISA for measurement of cytokine levels in co-culture supernatants
ELISA was used to measure the levels of cytokines interferon-gamma (IFN-γ; 70-EK180-48, Lianke, China), tumor necrosis factor-alpha (TNF-α; 70-EK182HS-48, Lianke, China), IL-10 (70-EK110/2-96, Lianke, China) in the supernatants. Standards, samples, and blanks were incubated (37°C, 1 h) and sequentially added with detection solutions A and B (37°C, 1 h and 30 min, respectively), TMB substrate (PR1210, Solarbio, Beijing, China), and stop solution. Absorbance (450 nm) was recorded by a microplate reader (CMax Plus, Molecular Devices Instruments Co., Ltd., USA), and cytokine concentrations were determined from a standard curve.
Cell migration assay
HCT116 cells were cultured in the lower chamber of a 24-well Transwell system (3421, Corning, USA), with 5 × 105 CAR-T cells (PD-1/Nb, FAP/Nb, or FAP-PD-1/Nb) added to the upper chamber. The chambers were incubated at 37°C in a 5% CO2 incubator for 24 h. The cells that migrated through the membrane were fixed, stained with crystal violet (1425163, Leagene, China) for 20 min, and enumerated by microscopy (https://www.bjcossim.com/p/m/bld-200/, BLD-200, Cossim, China).
Cell invasion assay
Tumor cell invasion was assessed with Matrigel-coated Transwell inserts (356234, Corning, USA) to simulate the tumor stroma barrier. FAP-PD-1/Nb CAR-T cells (or other subtypes) were seeded onto pre-polymerized Matrigel in the upper Transwell chamber. After a 24-h incubation (37°C, 5% CO2), non-invasive cells were removed, and invasive cells were stained with crystal violet (0.1%) for 20 min, followed by microscopic enumeration (https://www.bjcossim.com/p/m/bld-200/, BLD-200, Cossim, China).
Statistical analysis
Data analysis was performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). Results are presented as mean ± Standard deviation (n = 3). Independent sample t-test was used for comparisons between two groups, while a one-way analysis of variance followed by Tukey’s multiple comparison test was used for comparisons between three or more groups. Statistical significance was set at P < 0.05.
RESULTS
Successful construction of dual-targeting CAR-T cells targeting PD-L1 Nb and CAF
ELISA was employed to measure PD-1/Nb levels in the culture supernatants collected at various time points to confirm PD-1/Nb secretion by CAR-T cells. The findings indicated sustained PD-1/Nb release from PD-1/Nb CAR-T cells and FAP-PD-1/Nb CAR-T cells [Figure 1a]. WB assessed PD-1/Nb protein expression in PD-1/Nb CAR-T, FAP/Nb CAR-T, FAP-PD-1/Nb CAR-T, and UTD groups. PD-1/Nb was expressed in PD-1/Nb CAR-T and FAP-PD-1/Nb CAR-T cells but not in the UTD group. Notably, PD-1/Nb expression was significantly higher in FAP-PD-1/Nb CAR-T cells than in PD-1/Nb CAR-T cells (P < 0.01) [Figure 1b and c]. After co-culturing FAP-PD-1/Nb CAR-T cells with HCT116 and SW480 target cells, flow cytometry was employed to detect the expression of FAP on the cell surface. FAP was expressed in HCT116 and SW480 cells (P < 0.01) [Figure 1d and e]. These results confirmed the successful construction of dual-targeting CAR-T cells targeting PD-L1 Nb and CAF.

- Construction of dual-targeting CAR-T cells targeting PD-L1 Nb and CAF. (a) ELISA demonstrated sustained PD-1/Nb secretion from PD-1/Nb and FAP-PD-1/Nb CAR-T cells. (b and c) Western blot confirmed PD-1/Nb expression (✶✶P < 0.01, vs. UTD control). (d and e) Flow cytometry analysis of FAP expression in HCT116 and SW480 target cells following co-culture with FAP-PD-1/Nb CAR-T cells (✶✶P < 0.01, vs. HCT116 or SW480 group). n = 3. CAR-T: Chimeric antigen receptor T cell, ELISA: Enzyme-linked immunosorbent assay, CAF: Cancer-associated fibroblast, PD-L1: Programmed death-ligand 1, Nb: Nanobody, SD: Standard deviation, UTD: Untreated, FAP: fibroblast activation protein.
FAP activates CAR-T cells
CD25 and CD69 are key markers of T-cell activation. CAR-T cells were co-cultured with FAP-positive target cells for 72 h and subjected to flow cytometry analysis of CD25+CD69+ expression to assess the impact of FAP on CAR-T cell activation [Figure 2]. The percentage of CD25+CD69+ CAR-T cells increased sequentially in the PD-1/Nb, FAP/Nb, and FAP-PD-1/Nb groups, with the FAP-PD-1/Nb group exhibiting significantly higher activation (P < 0.01). Hence, FAP-PD-1/Nb CAR-T cells exhibited enhanced activation in response to FAP-expressing target cells.
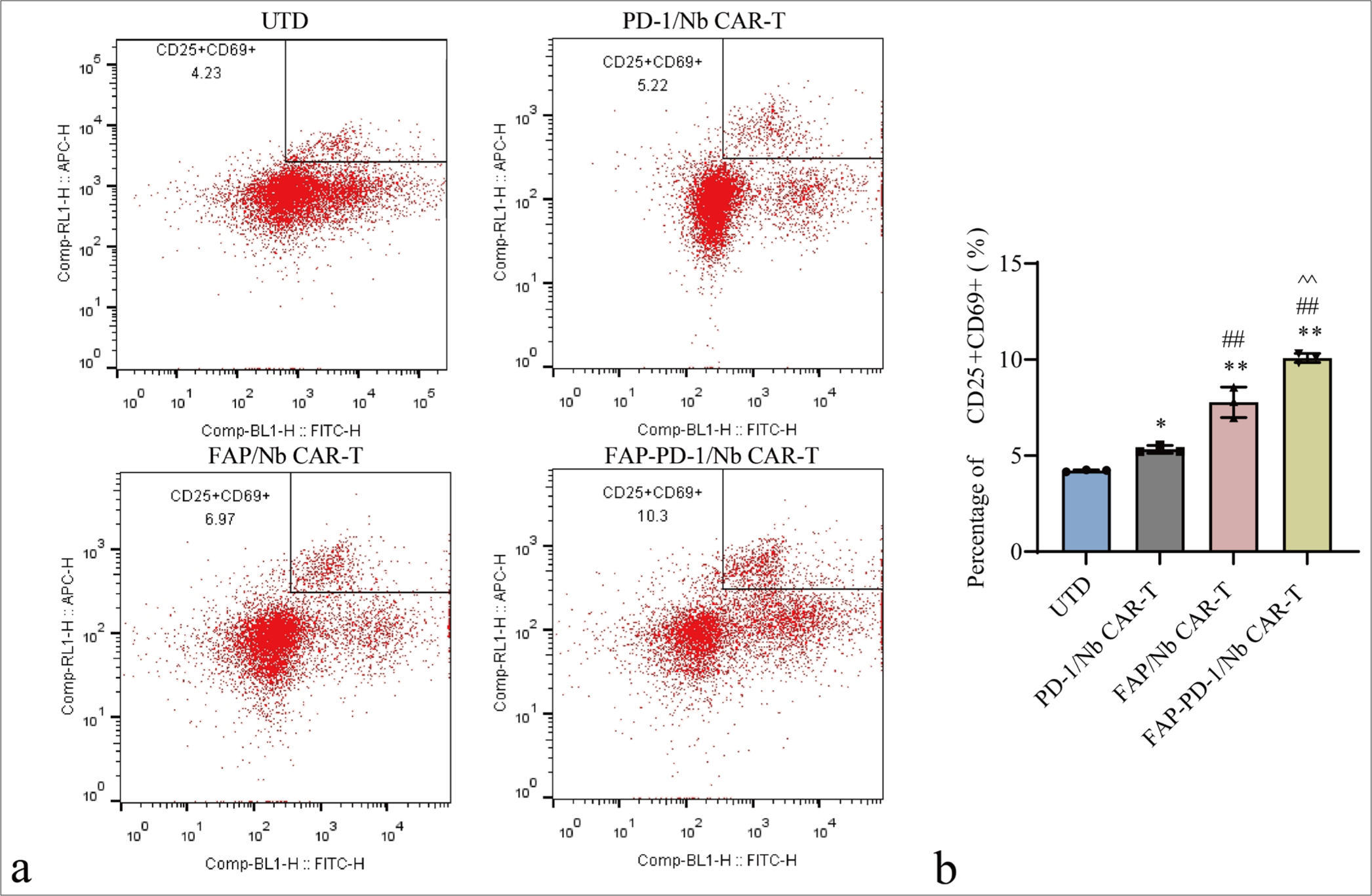
- Flow cytometry analysis of CD25+CD69+ level on the surface of CAR-T cells. (a) Representative flow cytometry plots showing the expression of CD25+CD69+ on the surface of CAR-T cells. (b) Quantification of CD25+CD69+ levels. n = 3. ✶P < 0.05, ✶✶P < 0.01 (vs. UTD control). ##P < 0.01 (vs. PD-1/Nb CAR-T). ^^P < 0.01 (vs. FAP/Nb CAR-T). CAR-T: Chimeric antigen receptor T cell, CD: Cluster of differentiation, UTD: Untreated, Nb: Nanobody, PD-1: Programmed cell death protein 1, FAP: Fibroblast activation protein.
FAP-PD-1/Nb CAR-T cells exhibit high killing activity against FAP-positive CRC cell lines in vitro
Apoptosis in HCT116 cells following co-culture with various CAR-T cell types was assessed. The proportion of apoptotic target cells in the FAP-PD-1/Nb CAR-T cell group was significantly higher than that in the PD-1/Nb CAR-T and FAP/Nb CAR-T cell groups (P < 0.01) [Figure 3a-d], further confirming the killing effect of dual-targeting CAR-T cells. CCK-8 assays revealed significantly greater proliferation inhibition of HCT116 cells by FAP-PD-1/Nb CAR-T cells compared with the other groups (P < 0.01) [Figure 3e], further confirming the anti-tumor effect of dual-targeting CAR-T cells.
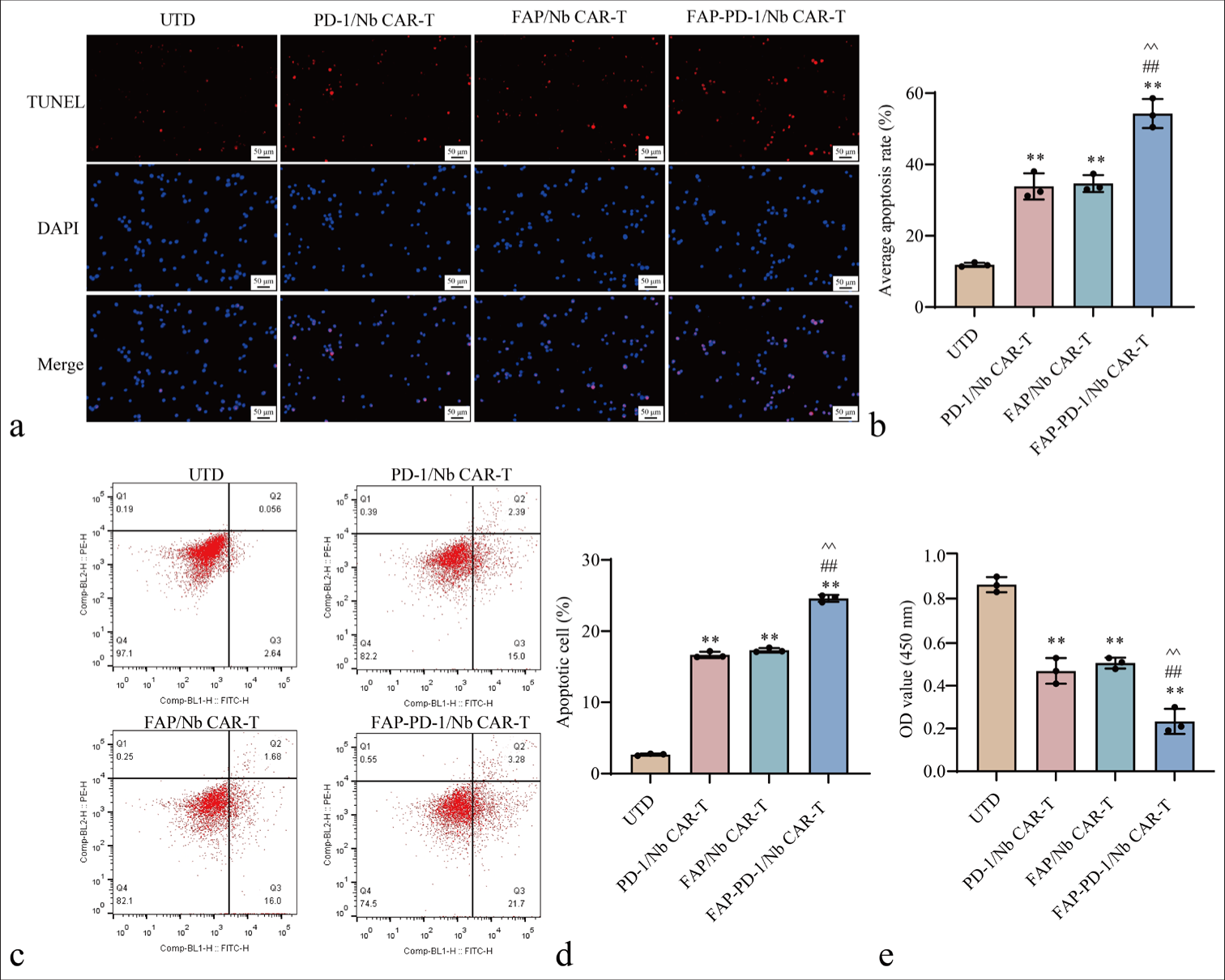
- Effects of FAP-PD-1/Nb CAR-T cells on FAP-positive CRC cells. (a and b) TUNEL assay assessing apoptosis in HCT116 cells. (c and d) Flow cytometry analysis determining apoptosis. (e) CCK-8 assay evaluating proliferation in HCT116 cells (n = 3; ✶✶P < 0.01 vs. UTD; ##P < 0.01 vs. PD-1/Nb CAR-T; ^^P < 0.01 vs. FAP/Nb CAR-T). n = 3. CAR-T: Chimeric antigen receptor T cell, TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling, CCK-8: Cell counting kit-8, FAP: Fibroblast activation protein, UTD: Untreated, Nb: Nanobody, PD-1: Programmed cell death protein 1, CRC: Colorectal cancer.
Transwell migration and invasion assays were conducted to evaluate the migratory and invasive potential of FAPPD-1/Nb CAR-T cells within the TME. FAP-PD-1/Nb CAR-T cells exhibited significantly greater migration than PD-1/Nb CAR-T cells, FAP/Nb CAR-T cells, and UTD control (P < 0.01) [Figure 4a and b]. Similarly, FAP-PD-1/Nb CAR-T cells showed notably enhanced invasiveness compared with single-target CAR-T cells and UTD control (P < 0.01) [Figure 4c and d]. This finding suggested that FAP-PD-1/Nb CAR-T cells effectively migrated toward the tumor site and exhibited enhanced permeability, enabling them to efficiently penetrate the extracellular matrix barrier.
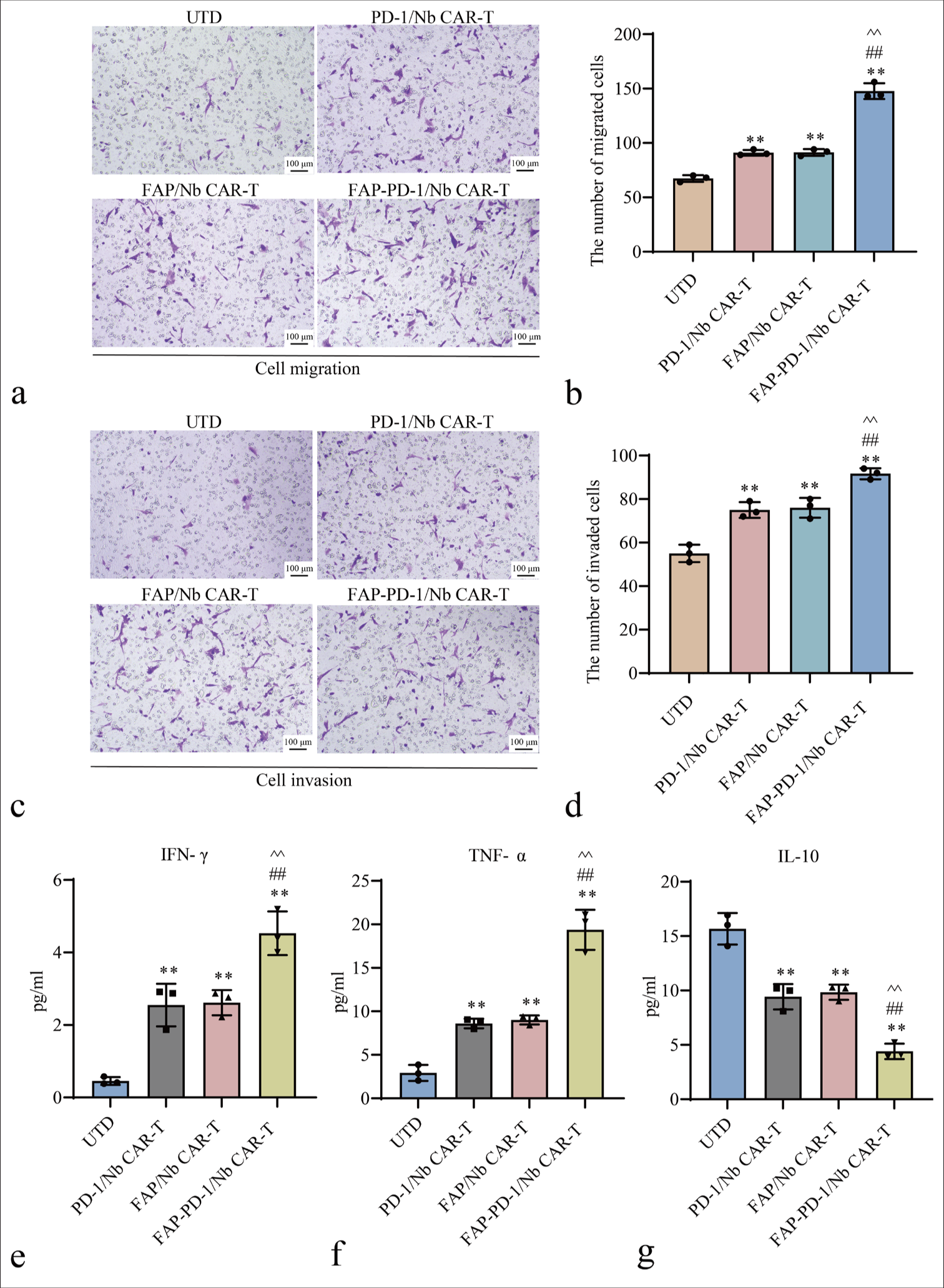
- Evaluation of cell migration, invasion capacities, and cytokine in different CAR-T Treatment Groups. (a and b) Transwell migration assay results for different CAR-T cell types. (c and d) Matrigel invasion assay results for different CAR-T cell types. (e-g) ELISA analysis of IFN-γ, TNF-α, and IL-10 levels (✶✶P < 0.01 vs. UTD; ##P < 0.01 vs. PD-1/Nb CAR-T; ^^P < 0.01 vs. FAP/Nb CAR-T). n = 3. CAR-T: Chimeric antigen receptor T cell, ELISA: Enzyme-linked immunosorbent assay, UTD: Untreated, Nb: Nanobody, PD-1: Programmed cell death protein 1, FAP: Fibroblast activation protein, IFN-γ: Interferon-gamma, TNF-α: Tumor necrosis factor-alpha, IL-10: Interleukin-10.
FAP-PD-1/Nb CAR-T cells effectively regulate the cytokine network
The supernatant levels of IFN-γ, TNF-α, and IL-10 were measured by ELISA following co-culture of CAR-T cells with HCT116 cells. FAP-PD-1/Nb CAR-T cells showed significantly higher IFN-γ and TNF-α and significantly lower IL-10 than PD-1/Nb and FAP/Nb CAR-T cells (P < 0.01, Figure 4e-g). Hence, dual-targeting CAR-T cells effectively regulated the cytokine network and promoted anti-tumor immune responses.
FAP-PD-1/Nb CAR-T Cells Show a Certain Level of Safety
Flow cytometry was used to assess the cytotoxicity of FAP-PD-1/Nb CAR-T cells against PBMCs at various concentrations. Dual-targeting CAR-T cells had a low killing rate on PBMCs, with no significant differences observed when compared with the UTD group (P > 0.05), suggesting that FAP-PD-1/Nb CAR-T cells had minimal toxicity toward PBMCs [Figure 5a and b]. Furthermore, TUNEL and CCK-8 assays indicated no significant changes in PBMC apoptosis or viability following co-culture with FAP-PD-1/Nb CAR-T cells when compared with the UTD group [Figure 5c-e]. These findings indicated that FAP-PD-1/Nb CAR-T cells had a certain level of safety.
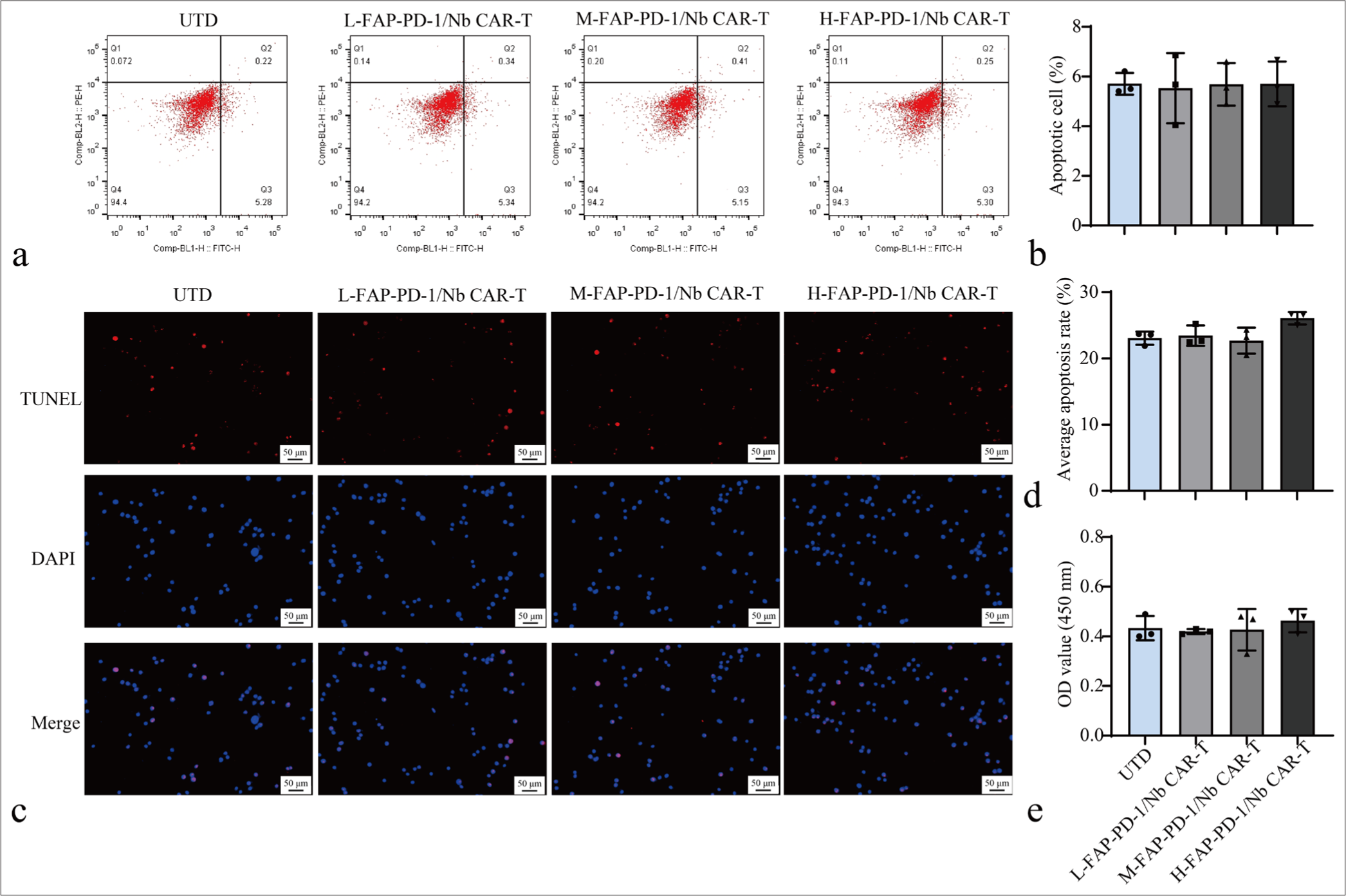
- FAP-PD-1/Nb CAR-T Cells safety evaluation. (a and b) Flow cytometry analysis of apoptosis in PBMCs co-cultured with different concentrations of different CAR-T cell types. (c and d) TUNEL assay for detecting cell apoptosis. (e) CCK-8 assay for detecting cell proliferation. Data are representative of three independent experiments. n = 3. CAR-T: Chimeric antigen receptor T cell, PBMC: Peripheral blood mononuclear cell, TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling, CCK-8: Cell counting kit-8, Nb: Nanobody, FAP: Fibroblast activation protein, PD-1: Programmed cell death protein 1.
DISCUSSION
Recent advances in cancer immunotherapy, particularly CAR T-cell therapy, offer hope for improved treatment outcomes. Nevertheless, immunosuppressive components within the tumor milieu restrict the therapeutic potential of CAR T cells.[15] This study aimed to address these issues by constructing dual-targeting CAR-T cells targeting PD-L1 Nb and CAF. Our main findings demonstrate the successful construction of FAP-PD-1/Nb CAR-T cells, which not only effectively recognize and kill FAP-positive CRC cells but also secrete PD-1 Nb, thereby overcoming PD-L1-mediated immune escape and exhibiting high anti-tumor activity and safety.
CAR T-cell therapy is still in its infancy and faces considerable hurdles. A case study involving CAR-T cell therapy for patients with ERBB2-overexpressing metastatic CRC reported one patient who developed rapid respiratory distress after receiving the treatment and died within 5 days. Further investigation revealed that CAR-T cells may have attacked lung epithelial cells, leading to acute lung injury. This case highlights the potential for off-target effects with CAR T-cell therapy, underscoring the need for further safety research.[16] The current research on CAR T-cell immunotherapy for solid tumors, including CRC, focuses on identifying optimal target antigens to improve treatment efficacy. Promising targets under investigation in CRC include carcinoembryonic antigen, epidermal growth factor receptor, epithelial cell adhesion molecule, and tumor-associated glycoprotein-72.[17] In recent years, PD-1 has attracted widespread attention. It is a transmembrane receptor on T lymphocytes, which was first obtained from an apoptotic mouse T lymphocyte hybridoma through subtractive hybridization. This protein (PD-1), which was named for its role in apoptosis, is primarily engaged by its ligand, PD-L1.[18] PD-L1 mediates immune evasion through PD-1-dependent T-cell suppression and is overexpressed in a range of solid tumors, including melanoma, lung, pancreatic, and CRC.[19-22] Therefore, PD-L1 can also be a new target for CAR-T treatment. The proposed combination therapy of PD-1 antibodies and CAR-T technology has been tested in immunodeficient mice, thereby validating the feasibility of this approach.[23]
FAP serves as a specific marker for CAFs and has high expression in TME but minimal expression in normal tissues and fibroblasts.[24,25] The role of FAP in tumorigenesis is increasingly recognized, with evidence suggesting its involvement in extracellular matrix degradation, angiogenesis, tumor cell proliferation, and metastasis.[26] Overexpression of FAP in various cancers, including liver, gliomas, and CRCs, correlates with poor prognosis.[27-29] Shahvali et al.[30] demonstrated the efficacy of FAP-targeted CAR T-cells in recognizing and eliminating FAP-expressing cells and reported their use as a promising therapeutic strategy against CAFs and solid tumors.
Nbs are derived from the single heavy chain of antibodies found in the serum of camelid animals (such as camels and alpacas).[31] Nbs exhibit enhanced targeting capability, reduced immunogenicity, and superior stability and thermal resilience compared with traditional full-length antibodies (e.g., IgG).[32] In CAR-T cells, Nbs, as antigen recognition components, can simplify the CAR engineering process without complex modifications CARs with different affinities can be constructed to optimize treatment efficacy and reduce side effects.[33] Xie et al. reported that CAR T cells engineered with Nbs targeting murine CD47 and secreting PD-L1 exhibited enhanced in vivo proliferation.[34] We constructed novel CAR-T cells that target FAP and secrete PD-1 Nb based on FAP Nb and PD-1 Nb to explore their application value in solid tumors and provide new ideas for CAR-T cell therapy. This study validates the hypothesis that bispecific FAP-PD-1/Nb CAR-T cells augment anti-tumor immunity. Of the three CAR-T cell groups (PD-1/Nb, FAP/Nb, and FAP-PD-1/Nb), only FAP-PD-1/Nb cells consistently released PD-1 Nb and blocked PD-L1-driven immune suppression.
Flow cytometry assessed the surface expression of the early activation markers CD25 and CD69 to confirm the enhanced activation of FAP-PD-1/Nb CAR-T cells.[35] Stimulation by target cells increased CD25 and CD69 expression in CAR T cells, in contrast to unstimulated CAR T cells or normal T cells.[36] In the present research, flow cytometry revealed significantly higher CD25 and CD69 expression in FAPPD-1/Nb CAR-T cells compared with the other groups. The results also verified the importance of CAF in promoting CAR-T cell stimulation and indicated that this dual-targeting CAR-T cell strategy effectively enhanced T cell stimulation and immune responses. In in vitro functional experiments, FAP-PD-1/Nb CAR-T cells exhibited high killing efficiency and anti-tumor effects, especially in FAP-positive HCT116 cell lines. Furthermore, FAP-PD-1/Nb CAR-T cells exhibited notably greater cytotoxicity and proliferation inhibition against target cells compared with single-target CAR-T cells. In addition, FAP-PD-1/Nb CAR-T cells showed superior migration and invasion abilities compared with the single-target CAR-T cell group and the UTD group. These findings suggest that FAP-PD-1/Nb CAR-T cells not only possess potent anti-tumor activity but also exhibit enhanced capability to traverse the TME and infiltrate tumor tissues, thereby improving their therapeutic efficacy.
A retrospective study indicates that CAR T-cell therapy might enhance anti-tumor immunity by increasing IFN-γ and TNF-α levels. Improved CAR T-cell treatment outcomes could correlate with elevated levels of these cytokines.[37] Our study demonstrates that FAP-PD-1/Nb CAR-T cells significantly enhanced the secretion of IFN-γ and TNF-α while simultaneously reducing the levels of the immunosuppressive cytokine IL-10. Hence, FAP-PD-1/Nb CAR-T cells not only exhibit excellent cytotoxicity but also enhance anti-tumor immunity by modulating the immune microenvironment. These changes in the cytokine network may facilitate T-cell activation, inhibition of immune escape, and tumor cell clearance. Through these mechanisms, FAP-PD-1/Nb CAR-T cells may effectively alter the immunosuppressive state within the TME, thereby significantly improving therapeutic outcomes. Consistent with the findings of other researchers, the present study demonstrates that the dual targeting strategy engaging CAFs and PD-L1 can improve CAR T-cell therapy efficacy.[38] For example, previous research demonstrated that CAFs suppress immune effector cell function within the TME, and targeting CAFs can substantially improve immunotherapy efficacy.[39] Furthermore, the therapeutic efficacy of PD-1/PD-L1 blockade has been demonstrated in numerous cancer types. Our study further confirms that PD-1 Nb secretion mediated by CAR-T cells can directly act in the TME, thereby enhancing immune responses. Engineered CAR-T cells can damage normal tissues to varying degrees while killing tumor cells.[40] Our results show that FAP-PD-1/Nb CAR-T cells have a low killing rate on normal PBMCs in vitro and have no significant effect on PBMC apoptosis and viability, indicating good safety.
This study employed dual-targeting CAR-T cells, which innovatively target the tumor immune escape and the supporting structure of the TME and CAFs, thereby effectively addressing the constraints of single-targeting CAR-T cells in solid tumor therapy. This study primarily used FAP-positive CRC cell lines (HCT116 and SW480); therefore, the generalizability of these findings to other cancer types requires further investigation. In practical clinical applications, CAR-T cells may exert off-target toxicity or long-term side effects on normal tissues. Therefore, evaluating the potential long-term effects and off-target toxicity of FAP-PD-1/Nb CAR-T cells is a crucial step to ensure their safety. Although the results of the in vitro experiments show good anti-tumor activity, they were not verified using in vivo models, thereby limiting the extrapolability of the results. Species differences are another factor that may affect the results. While in vitro data are informative, the complexity of the in vivo immune system suggests that the performance of FAP-PD-1/Nb CAR-T cells in animal models or clinical settings may differ from in vitro observations. Therefore, future studies should further assess the potential toxicity and off-target effects of FAP-PD-1/Nb CAR-T cells in non-tumor tissues expressing FAP by using mouse models with a complete immune system. Long-term follow-up studies should also be conducted to evaluate the persistence of FAP-PD-1/Nb CAR-T cells, their impact on the immune system, and whether they induce chronic immune responses or other side effects. Future research can explore how to improve the persistence of dual-targeting CAR-T cells and their infiltration capacity in the TME and further evaluate their safety and long-term anti-tumor effects in in vivo models. We should also explore the potential for combined applications with other treatment modalities (such as radiotherapy, chemotherapy, or immune checkpoint inhibitors) to achieve effective treatment regimens.
Future research will utilize a mouse xenograft model to assess the anti-tumor efficacy of FAP-PD-1/Nb CAR-T cells and evaluate their potential toxicity and off-target effects on non-tumor tissues expressing FAP. To further investigate the long-term effects, we plan to regularly collect serum samples to measure immune biomarkers (such as cytokines and antibody levels) and perform necropsy and histological examinations to assess the overall health status post-treatment. This strategy will allow us to evaluate the persistence of FAP-PD-1/Nb CAR-T cells, their impact on the immune system, and whether they induce chronic immune responses or other adverse effects. Future studies could also explore strategies to enhance the persistence and infiltration capacity of dual-targeted CAR-T cells within the TME as well as assess their safety and long-term anti-tumor effects in vivo. At the same time, the potential for combination with other therapeutic approaches (such as radiotherapy, chemotherapy, or immune checkpoint inhibitors) should be explored to develop effective treatment regimens. Although Nbs exhibit lower immunogenicity compared with traditional antibodies, immunogenicity remains a key issue in their clinical application. A series of immunogenicity assessments will be planned, particularly a long-term immune tolerance study following repeated administration in animal models. Humanized or engineered Nbs will be employed to minimize their immunogenicity in humans. Furthermore, to improve their feasibility in clinical applications, we plan to evaluate the efficacy of different Nb sequences to identify those with optimal immune tolerance and therapeutic efficacy.
SUMMARY
This study successfully developed dual-targeting FAPPD-1/Nb CAR-T cells, which demonstrated potent anti-tumor activity. These CAR-T cells effectively targeted CAFs and PD-L1, thereby significantly enhancing tumor cell killing and proliferation inhibition in FAP-positive CRC cell lines. They also promoted immune activation by increasing the secretion of IFN-γ and TNF-α while reducing IL-10. Importantly, FAP-PD-1/Nb CAR-T cells showed minimal cytotoxicity to PBMCs, indicating a favorable safety profile. This dual-targeting approach offers a promising therapeutic strategy for CRC in clinical application. However, several challenges remain before clinical translation, including the high cost and complexity of CAR-T cell production, precision in patient selection, tumor heterogeneity, and immune escape issues. Future efforts will need to focus on optimizing therapeutic strategies and addressing these challenges for successful clinical application.
ACKNOWLEDGMENTS
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The datasets and materials generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
ABBREVIATIONS
293T: Human embryonic kidney 293 cells
APC: Allophycocyanin
BCA: Bicinchoninic Acid protein assay
CAFs: Cancer-Associated Fibroblasts
CAR: Chimeric Antigen Receptor
CCK-8: Cell Counting Kit-8
CD25: Cluster of Differentiation 25
CD69: Cluster of Differentiation 69
CRC: Colorectal cancer
DAPI: 4’,6-Diamidino-2-Phenylindole
EDTA: Ethylenediaminetetraacetic Acid
ELISA: Enzyme-Linked Immunosorbent Assay
FAP: Fibroblast Activation Protein
FITC: Fluorescein Isothiocyanate
GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase
GFP: Green Fluorescent Protein
GV400: Lentiviral vector
His-tag: Histidine tag
HRP: Horseradish Peroxidase
IFN-γ: Interferon-gamma
IgG: Immunoglobulin G
IL-10: Interleukin-10
IL-2: Interleukin-2
MHC: Major Histocompatibility Complex
Nb: Nanobody
OD: Optical Density
PBMCs: Peripheral Blood Mononuclear Cells
PBS: Phosphate-Buffered Saline
PCR: Polymerase Chain Reaction
PD-1: Programmed Death-1
PD-L1: Programmed Death-Ligand 1
PE: Phycoerythrin
RPMI: Roswell Park Memorial Institute medium
SDS-PAGE: Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
STR: Short Tandem Repeat
TBST: Tris-Buffered Saline with Tween 20
TMB: 3,3’,5,5’-Tetramethylbenzidine
TME: Tumor Microenvironment
TNF-α: Tumor Necrosis Factor-alpha
Transwell: Transwell chamber
TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling
Western blot: Western blot analysis
AUTHOR CONTRIBUTIONS
YG and CJL: Contributed to the study design and literature research, and participated in manuscript preparation; HY: Contributed to manuscript editing; QJX: Contributed to data acquisition; HJH: Guarantor the integrity of the entire study; JWL and JDM: Contributed to the study concepts; YG: Defined the intellectual content and was involved in manuscript review.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval and consent were not applicable for this study as it did not involve human participants, animal experiments, or clinical trials. The data and materials used were obtained through laboratory-based experiments with no involvement of human subjects or living organisms.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: Not applicable.
References
- Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209-49.
- [CrossRef] [PubMed] [Google Scholar]
- A systematic review of the role of chimeric antigen receptor T (CAR-T) cell therapy in the treatment of solid tumors. Cureus. 2021;13:e14494.
- [CrossRef] [Google Scholar]
- An update on colorectal cancer. Curr Probl Surg. 2018;55:76-116.
- [CrossRef] [PubMed] [Google Scholar]
- Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332-7.
- [CrossRef] [PubMed] [Google Scholar]
- m(6) A methylation modification and immune cell infiltration: implications for targeting the catalytic subunit m(6)A-METTL complex in gastrointestinal cancer immunotherapy. Front Immunol. 2023;14:1326031.
- [CrossRef] [PubMed] [Google Scholar]
- Tumor necrosis factor receptor 2/AKT and ERK signaling pathways contribute to the switch from fibroblasts to CAFs by progranulin in microenvironment of colorectal cancer. Oncotarget. 2017;8:26323-33.
- [CrossRef] [PubMed] [Google Scholar]
- CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021;11:69.
- [CrossRef] [PubMed] [Google Scholar]
- Blocking CD30 on T cells by a dual specific CAR for CD30 and colon cancer antigens improves the CAR T cell response against CD30(-) Tumors. Mol Ther. 2019;27:1825-35.
- [CrossRef] [PubMed] [Google Scholar]
- Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059-70.
- [CrossRef] [PubMed] [Google Scholar]
- Anti-PD-1/PD-L1 therapy for colorectal cancer: Clinical implications and future considerations. Transl Oncol. 2024;40:101851.
- [CrossRef] [PubMed] [Google Scholar]
- PD-1/PD-L1 inhibitors for early and middle stage microsatellite high-instability and stable colorectal cancer: A review. Int J Colorectal Dis. 2024;39:83.
- [CrossRef] [PubMed] [Google Scholar]
- FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem Biophys Res Commun. 2017;487:8-14.
- [CrossRef] [PubMed] [Google Scholar]
- Research on anti-tumor effect and mechanism of the new CARTCells secreting PD-1 nanobody targeting FAP [Doctor] China: Guangxi Medical University; 2020. p. :51-82.
- [Google Scholar]
- Colorectal cancer cell intrinsic fibroblast activation protein alpha binds to Enolase1 and activates NF-κB pathway to promote metastasis. Cell Death Dis. 2021;12:543.
- [CrossRef] [PubMed] [Google Scholar]
- Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. 2022;12:6273-90.
- [CrossRef] [PubMed] [Google Scholar]
- Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843-51.
- [CrossRef] [PubMed] [Google Scholar]
- Updated clinical perspectives and challenges of chimeric antigen receptor-T cell therapy in colorectal cancer and invasive breast cancer. Arch Immunol Ther Exp (Warsz). 2023;71:19.
- [CrossRef] [PubMed] [Google Scholar]
- Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 2018;13:e0198347.
- [CrossRef] [PubMed] [Google Scholar]
- PD-1, PD-L1, and PD-L2 gene expression and tumor infiltrating lymphocytes in canine melanoma. Vet Pathol. 2021;58:692-8.
- [CrossRef] [PubMed] [Google Scholar]
- A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017;10:2115-26.
- [CrossRef] [PubMed] [Google Scholar]
- Prognostic and clinicopathological value of PD-L1 in colorectal cancer: A systematic review and meta-analysis. Onco Targets Ther. 2019;12:3671-82.
- [CrossRef] [PubMed] [Google Scholar]
- Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2:e26286.
- [CrossRef] [PubMed] [Google Scholar]
- The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020;39:783-803.
- [CrossRef] [PubMed] [Google Scholar]
- Fibroblast activation protein overexpression and clinical implications in solid tumors: A meta-analysis. PLoS One. 2015;10:e0116683.
- [CrossRef] [PubMed] [Google Scholar]
- Fibroblast activation protein. Cancer Biol Ther. 2012;13:123-9.
- [CrossRef] [PubMed] [Google Scholar]
- Fibroblast activation protein promotes progression of hepatocellular carcinoma via regulating the immunity. Cell Biol Int. 2024;48:577-93.
- [CrossRef] [PubMed] [Google Scholar]
- The expression of fibroblast activation protein in human colon carcinoma. J Clin Oncol. 2005;23:9668.
- [CrossRef] [Google Scholar]
- Stromal expression of fibroblast activation protein alpha (FAP) predicts platinum resistance and shorter recurrence in patients with epithelial ovarian cancer. Cancer Microenviron. 2015;8:23-31.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting fibroblast activation protein (FAP): Advances in CAR-T cell, antibody, and vaccine in cancer immunotherapy. Drug Deliv Transl Res. 2023;13:2041-56.
- [CrossRef] [PubMed] [Google Scholar]
- Unveiling the potential of extracellular vesicles as biomarkers and therapeutic nanotools for gastrointestinal diseases. Pharmaceutics. 2024;16:567.
- [CrossRef] [PubMed] [Google Scholar]
- Nanobody-A versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021;13:e1697.
- [CrossRef] [PubMed] [Google Scholar]
- The application of nanobody in CAR-T therapy. Biomolecules. 2021;11:238.
- [CrossRef] [PubMed] [Google Scholar]
- Improved antitumor efficacy of chimeric antigen receptor T cells that secrete single-domain antibody fragments. Cancer Immunol Res. 2020;8:518-29.
- [CrossRef] [PubMed] [Google Scholar]
- High-dimensional functional phenotyping of preclinical human CAR T cells using mass cytometry. STAR Protoc. 2022;3:101174.
- [CrossRef] [PubMed] [Google Scholar]
- Bispecific CAR-T cells targeting FAP and GPC3 have the potential to treat hepatocellular carcinoma. Mol Ther Oncol. 2024;32:200817.
- [CrossRef] [PubMed] [Google Scholar]
- Complex association of body mass index and outcomes in patients with relapsed and refractory multiple myeloma treated with CAR-T cell immunotherapy. Cytotherapy. 2024;26:832-41.
- [CrossRef] [PubMed] [Google Scholar]
- CAR T cells and checkpoint inhibition for the treatment of glioblastoma. Expert Opin Biol Ther. 2020;20:579-91.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting CAFs to improve Anti-PD-1 checkpoint immunotherapy. Cancer Res. 2023;83:655-6.
- [CrossRef] [PubMed] [Google Scholar]
- CAR-T cells neurotoxicity from consolidated practice in hematological malignancies to fledgling experience in CNS tumors: Fill the gap. Front Oncol. 2023;13:1206983.
- [CrossRef] [PubMed] [Google Scholar]




