


Translate this page into:
Exploring miR-3148’s impact on Krüppel-like factor 6-driven mitophagy and apoptosis in myocardial ischemic injury

Lei Xian

Ying He
*Corresponding authors: Lei Xian, Department of Thoracic and Cardiovascular Surgery, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China. efyxwk@163.com
Ying He The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China. Department of Pharmacy, The Fourth Affiliated Hospital of Guangxi Medical University, Liuzhou, Guangxi, China. 2028402005@st.gxu.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Huang C, Li L, Deng H, Su J, Wei Q, He Y, et al. Exploring miR-3148’s impact on krüppel-like factor 6-driven mitophagy and apoptosis in myocardial ischemic injury. CytoJournal. 2025;22:19. doi: 10.25259/Cytojournal_209_2024
Abstract
Objective
Myocardial infarction (MI) is a leading cause of death worldwide, accounting for millions of fatalities annually. The injury and repair of cardiomyocytes are closely associated with the changes in gene expression. MicroRNAs could serve as a potential target for MI treatment. This work aims to investigate the role of miR-3148 in mitochondrial dynamics during acute MI (AMI) with a specific focus on its regulatory mechanisms in mitophagy and apoptosis, which could reveal potential therapeutic targets for AMI treatment.
Material and Methods
MiR-3148 levels in patients with AMI and experimental models were measured to assess the effects of miR-3148 on cardiomyocyte viability under oxygen and glucose deprivation (OGD). The present investigation involved monitoring mitophagy markers, including PTEN-induced kinase 1 (PINK1), parkin RBR E3 ubiquitin-protein ligase (Parkin), Beclin1, and microtubule-associated protein 1A/1B light chain 3 II/I (LC3 II/I) ratio, as well as apoptotic markers such as cysteine-aspartic acid protease (Caspase) 9, Caspase 3, and cytochrome C (Cyt C). In addition, Krüppel-like factor 6 (KLF6) was examined as a target of miR-3148.
Results
MiR-3148 was significantly elevated in patients with AMI and models. MiR-3148 overexpression reduced cardiomyocyte viability, whereas miR-3148 knockdown protected against OGD injury. The inhibition of miR-3148 activated mitophagy, as shown by the increased PINK1, Parkin, Beclin1 levels, and LC3 II/I ratios, and reduced sequestosome 1 (p62), and apoptotic markers levels. MiR-3148 directly targeted KLF6, reducing its expression. The suppression of KLF6 aggravated OGD injury by disrupting PINK1/Parkin-mediated mitophagy and enhancing apoptosis. Attenuating KLF6 expression reversed the protective effects of miR-3148 inhibition, indicating reciprocal regulation.
Conclusion
In myocardial ischemic injury, miR-3148 modulates PINK1/Parkin-mediated mitophagy and apoptosis through KLF6 regulation. This finding highlights miR-3148 as a key factor in the pathogenesis of AMI and as a potential therapeutic target.
Keywords
Apoptosis
Krüppel-like factor 6
MiR-3148
Mitophagy
Myocardial infarction

INTRODUCTION
Ischemic heart disease is a serious threat to human health because of its high morbidity and mortality rates, with acute myocardial infarction (AMI) being a leading cause of death. Restoring myocardial blood supply promptly is the most effective intervention for mitigating myocardial apoptosis, which contributes to cardiac dysfunction and myocardial remodeling.[1] Numerous treatment strategies have been verified to have strong cardioprotective effects in laboratory settings. However, the clinical efficacy of these cardioprotective therapies in patients with AMI has been disappointing due to their inability to restore energy metabolism, which is a crucial factor.[2] Mitochondria acts as the primary energy generators for adenosine triphosphate production in cells, with the mitochondrial quality control system (MQC) playing a crucial role in maintaining the homeostasis and survival of myocardial cells.[3,4] MQC is an endogenous cellular protective mechanism that coordinates various processes, including biogenesis, fission, fusion, protein degradation, and mitophagy, to regulate and maintain mitochondrial homeostasis. Mitochondrial dysfunction has been established as a precursor to cell death.[5] External stimuli, including reactive oxygen species stress, nutrient deficiency, and cellular aging, can lead to the gradual accumulation of mitochondrial DNA mutations, resulting in decreased mitochondrial membrane potential and subsequent depolarization damage. Mitophagy is initiated to maintain mitochondrial and cellular homeostasis and prevent damaged mitochondria from causing further cellular damage. During this process, damaged mitochondria are encapsulated in autophagosomes and fused with lysosomes, leading to their degradation. Mitophagy mediates the clearance of damaged, aging, and dysfunctional mitochondria; however, during myocardial ischemia, mitochondrial dynamics are disrupted, impairing mitophagy and resulting in mitochondrial dysfunction,[6] as well as cellular dysfunction and apoptosis.[7] Therefore, maintaining mitochondrial quality and promoting cell survival is critical.
MicroRNAs (miRNAs) are small noncoding RNAs that play crucial roles in the regulation of gene expression without encoding proteins. They participate in diverse biological processes, including cell cycle control, apoptosis, and various developmental and physiological processes. The expression of multiple miRNAs can be influenced by oxidative stress. Conversely, miRNAs can modulate the expression of redox sensors after the key components of cellular antioxidants interact with proteasomes and influence the DNA repair system. Recently, the redox-sensitive miRNAs has been a remarkable increase; notable targets include nuclear factor erythroid 2-related factor 2, silent information regulator 1, and nuclear factor kappa-B. These miRNAs have emerged as important regulators in myocardial ischemia injury. Research suggests that circulating miRNAs have potential as biomarkers for myocardial infarction (MI).[8] As a member of the miRNA family, miR-3148 has been recognized for its involvement in various pathological and physiological processes, including tumorigenesis and chronic thromboembolic pulmonary hypertension.[9-11] However, reports on its role in MI are scarce. In clinical practice, serum levels of miR-3148 have been quantified by using the quantitative reverse transcription polymerase chain reaction (qRT-PCR) in patients with AMI, revealing a considerable decrease following percutaneous coronary intervention (PCI). Meanwhile, the target gene prediction software programs microRNA Target Prediction Database, microRNA Data Integration Portal, and TargetScan identified Krüppel-like factor 6 (KLF6) as a potential target gene of miRNA-3148.
KLF6, a member of the Krüppel-like factor family, is widely expressed across the various tissues and participates in processes such as cell differentiation, tissue and organ development, cell growth, apoptosis, tumorigenesis, angiogenesis, and vascular repair. Recent studies have shown that KLF6 is highly up-regulated during early renal ischemiareperfusion injury (I/R) and that its targeted inhibition suppresses apoptosis,[12] indicating that it is closely associated with I/R. Moreover, KLF6 can counteract the activation of the intrinsic apoptotic pathway mediated by cytochrome C (Cyt C), thereby regulating mitochondrial function.[13] In acute liver injury, KLF6 induces autophagy through a tumor protein 53-dependent transcriptional mechanism, promoting liver regeneration and repair.[14] However, the regulatory mechanisms of KLF6 in AMI remain unclear.
This study found that miR-3148 was significantly elevated in patients and mice with AMI. To further investigate the role of miR-3148 in AMI, we examined its effect on myocardial cell viability in vivo and in vitro.
MATERIAL AND METHODS
Ethical approval
The ethics committee (2023-KY <0918>) granted approval for all the experiments, which were conducted in accordance with the international ethical guidelines, the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the Declaration of Helsinki.[15]
Patient population
Written informed consent was obtained from all the participants before their involvement in this study, which was approved by the medical research ethics committee. Blood samples were collected from patients with AMI (n = 10) and healthy individuals (n = 10) at the hospital. The identification of AMI in our research adhered to the inclusion criteria specified by the World Health Organization’s Definition of Myocardial Infarction: The revised version from 2008 to 2009.[16]
Cell culture, oxygen, and glucose deprivation (OGD), and cell transfection
Experiments were conducted by using AC16 cells (human cardiomyocytes, STCC131011G, Servicebio, Wuhan, China) cultured with Dulbecco’s Modified Eagle Medium (C1199500BT, DMEM, GIBCO, USA) supplemented with 10% fetal bovine serum (10099-141, GIBCO, Billings, MT, USA) and 1% penicillin-streptomycin (C0222, Beyotime, Shanghai, China) at 37°C with 5% Carbon dioxide (CO2). Cell line authentication was performed by using short tandem repeat (STR) analysis. Following the manufacturer’s instructions, the cell line was tested for mycoplasma contamination and confirmed to be negative.
OGD
Our OGD model was established as previously described.[17] After reaching 80% confluency, the cells were rinsed twice with phosphate-buffered saline (PBS) and then cultured in glucose-free and serum-free DMEM medium within an anaerobic environment by using an anaerobic tank equipped with an AnaeroPack (C-1, Mitsubishi Gas Chemical Company, Inc., Japan). The cells were maintained in a CO2 incubator for 6 h.
Cell transfection
AC16 cells were overexpressed with 50 nM miR-3148 mimic (Invitrogen; Sense, 5’-UGGAAAACUGGUGUGUGCU U-3’; antisense, 5 ‘-GCACACACCAGUUUUUUCCAUU-’) along with negative control (sense, 5’-UUCUCCGAAC GUGUUCACGUTT-3’; antisense, 5’-ACGUGACACGU CGGAGAATT -3’ from GenePharma Co., Ltd., Shanghai, China). MiR-3148 was knocked down in AC16 cells by using a miR-3148 inhibitor at a concentration of 50 nM (5’-AAGCACACACCAGUUUUUCCA-3’) along with an equivalent NC (5’-CAGUACUUUUGUAGUACAA -3’). In addition, the small interfering RNA targeting KLF6 (siKLF6) was designed, and si-KLF6-negative control (NC) was used as the negative control. AC16 cells were cultured to 30–50% confluence and transfected by using Lipofectamine 2000 (11668019, Invitrogen, Carlsbad, CA, USA). Transfected cells were collected 48 h later for subsequent experiments.
Animal model and antagomir-3148 administration
Male C57BL/6 mice aged 6–8 weeks and weighing 20 ± 2 g purchased from the Center for Animal Experiments of Guangxi Medical University were used in our study. All animals were handled in accordance with the international ethical guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. As previously mentioned,[18] the ligation of the proximal left anterior descending coronary artery was ligated by using 5-0# Prolene suture to induce myocardial ischemia. The mice were randomly divided into five groups: Control, Sham, Model, miR-3148 antagomir, and miR-3148 antagomir NC. The miR-3148 antagomir NC and miR-3148 antagomir groups were administered either miR-3148 antagomir NC or miR-3148 antagomir (80 mg/kg body weight dissolved in sterile PBS) through intraperitoneal injections for three consecutive days before AMI induction. Subsequently, the mice were anesthetized with 2,2,2-tribromoethanol (T48402-5G, Sigma, USA) at a dose of 200 mg/kg. Their hearts and blood were obtained by using the cervical dislocation method for further utilization.
qRT-PCR
Total Ribonucleic Acid (RNA) was extracted in accordance with the manufacturer’s protocol by using the RNAqueous® Total RNA Isolation Kit (Thermo Fisher Scientific Inc., AM1912, Waltham, MA, USA). Reverse transcription was performed by using the PrimeScriptTM room temperature (RT) reagent Kit (RR047A, Takara Biomedical Technology Co., Ltd., Osaka, Japan) for KLF6, PTEN-induced kinase 1 (PINK1) and parkin RBR E3 ubiquitin-protein ligase (Parkin) analysis. The GoTaq quantitative polymerase chain reaction (qPCR) Master Mix Kit (A6001, Promega Corporation, Madison, WI) was utilized to measure the messenger RNA (mRNA) levels of KLF6, PINK1, and Parkin genes. Mixing the GoTaq® qPCR Master Mix (×2), polymerase chain reaction (PCR) primers (×20) and DNA template. Nuclease-free water was then added to a final volume of 20 μL. A Real-Time PCR System (AriaMx, Agilen, California, USA) was used. The cycling parameters were as follows: Standard Cycling Conditions Steps: GoTaq® Hot Start Polymerase activation at 95°C for 2 min, 1 cycle. Denaturation was performed at 95°C for 15 s, followed by annealing and extension at 60°C for 1 min for 40 cycles. Before detection, centrifugation was performed briefly to collect the contents at the bottom of the wells. Differences in gene expression were calculated by using the relative quantitative 2−ΔΔCt method. β-actin was employed as the reference gene. For the analysis of miR-3148 expression, total RNA was reverse transcribed by using miRNA ALL-In-One complementary DNA (cDNA) Synthesis Kit (G898, ABM Inc., Zhenjiang, China), and miR-3148 levels were then detected with GoTaq qPCR Master Mix Kit. U6 served as the control. The primer specific for human miR-3148 and a universal 3’ miRNA reverse primer were procured from Applied Biological Materials Inc., Richmond, Canada. The other primers used were: KLF6 (human) forward 5’-GGCCAAGTTTACCTCCGACC-3’, reverse 5’-CGCAACCCCACAGTTGAGAA-3’, PINK1 (human) forward 5’-TGTCAGGAGATCCAGGCAATT-3’, reverse 5’-ATACTCCTCCAGCCGAAAGC-3’, Parkin (human) forward 5’-CAACAAATAGTCGGAACATC-3’, reverse 5’-GAACAAACTGCCGATCATTG-3’, miR-3148 (human) forward 5’- TGGAAAAAACTGGTGTGTGCTT-3’, U6 (human) forward 5’-CTCGCTTCGGCAGCACA-3’, reverse 5’-AAAATATGGAACGCTTCACG-3’, β-actin (human) forward 5’- AGTCATTCCAAATATGAGATGCGTT-3’ and reverse 5’-TGCTATCACCTCCCCTGTGT-3’.
Western blot (WB) assay
Tissues or cells were placed on ice boxes and then added with 200–400 uL of radio immunoprecipitation assay lysis buffer (RIPA) (R0010, Solarbio, Beijing, China) containing 1% phenylmethanesulfonyl fluoride (PMSF) (P6730, Solarbio, Beijing, China). The samples were subjected to grinding or pipetting for cell lysis, followed by homogenization, with the samples returned to the ice box every 5 s, then centrifuged 4℃ and 12000 rpm for 20 min (LX-165T2R, Haier Biomadical, Shanghai, China). A bicinchoninic acid (BCA) protein quantification kit (PC0020, Solarbio, Beijing, China) was used to determine the absorbance at 562 nm by using a microplate (FilterMax F3, MD, USA). Protein concentration was calculated in accordance with a standard curve. Protein blot analysis was conducted, and an equal amount of protein lysate was fractionated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred onto a polyvinylidene fluoride membrane. Following transfer, the membranes were blocked at RT for 20 min in protein-free fast-blocking buffer (×5) (PS108, EpiZyme, Shanghai, China) diluted to ×1 in Tris-buffered saline with Tween 20. Subsequently, the membranes were incubated overnight at 4°C with the following antibodies: Glyceraldehyde 3-phosphate dehydrogenase (1:1000, cat. no. ab8245; Abcam, USA), β-actin monoclonal antibody (β-actin; 1:20000, 66009-1-Ig, Proteintech, Wuhan, China), KLF6 (1:500, 14716-1-AP, Proteintech, Wuhan, China), PINK1 (1:1000, 23274-1-AP, Proteintech, Wuhan, China), PARK2/Parkin (1:2000, 14060-1-AP, Proteintech, Wuhan, China), microtubule-associated proteins 1A/1B light chain 3B (LC3)B (1:3000, ab192890, Abcam, USA), anti-sequestosome 1 (p62, 1:1000, K002179P, Solarbio, Beijing, China), Beclin1 (1:1000, K101553P, Solarbio, Beijing, China), cysteine-aspartic acid protease (Caspase) 3 (1:1000, K003262P, Solarbio, Beijing, China), Caspase 9 (1:1000, K109226P, Solarbio, Beijing, China), and Cyt C (1:1000, K107696P, Solarbio, Beijing, China). After being washed in Tris-buffered saline with Tween 20, the membranes were incubated with horseradish peroxidaseconjugated AffiniPure goat anti-rabbit Immunoglobulin G (IgG) (H + L) (1:10000, SA00001-2, Proteintech, Wuhan, China) or horseradish peroxidase-conjugated AffiniPure goat antimouse IgG (H + L) (1:10000, SA00001-1, Proteintech, Wuhan, China), at RT for 2 h. Afterward, the membranes were washed thrice and signals were detected by employing enhanced chemiluminescence reagents (BeyoECL Star, P0018AS, Beyotime, Shanghai, China). A gel imaging system (Tanon 5200, Tanon, Shanghai, China) was utilized to take photos, and ImageJ was used for density analysis.
Hematoxylin and eosin (H&E) staining
The myocardial tissues of mice were dehydrated by using a gradient technique, embedded in paraffin, and sliced into 5 μm sections. Subsequently, the sections were dewaxed then placed in 100% ethanol (200901, Yulu Experimental Equipment Co., LTD, Nanchang, China), 95% ethanol (191101, Yulu Experimental Equipment Co., LTD, Nanchang, China), 85% ethanol (190604, Yulu Experimental Equipment Co., LTD, Nanchang, China), and 75% ethanol (200801, Yulu Experimental Equipment Co., LTD, Nanchang, China) for 5 min each time and rinsed with pure water for 2 min. The sections were stained with hematoxylin (191202, Yulu Experimental Equipment Co., LTD, Nanchang, China) for 5 min with hydrochloric acid alcohol differentiation solution for 1–3 s and with eosin (200901, Yulu Experimental Equipment Co., LTD, Nanchang, China) for 1 min. They were then placed in 75% ethanol for 1 min, 85% ethanol for 1 min, 95% ethanol for 1 min, and 100% ethanol and dehydrated for 1, 5, and 5 min. Xylene permeation was conducted 3 times for 10 min each time. The sections were sealed with neutral gum and photographed under a light microscope (BX-53, Olympus, Japan).
Luciferase assay
GP-miRGLO-KLF6-3’-untranslated region (3’-UTR)-wild type (WT) and GP-miRGLO-KLF6-3’-UTR-mutant (MUT) luciferase reporter plasmids were purchased from Sangon Biotech (Shanghai, China). AC16 cells were seeded at a density of 1 × 105 cells per well in a 24-well cell culture plate. After 24 h of incubation, the cells were cotransfected with luciferase reporter plasmids (2 μg) and miR-3148 mimics (2 μg) by using Lipo2000 transfection reagent (BL623B, Biosharp, Anhui, China). A Dual-Luciferase Assay Kit (E2920, Promega Corporation, Madison, WI) was utilized to determine luciferase activity. A chemiluminescent enzyme-labeled instrument (GloMax 20/20, Promega Corporation, Madison, WI) was employed to quantify the luminescence signals of Firefly and Renilla. The Firefly/Renilla ratio served as an indicator for luciferase activity analysis.
Flow cytometry
Apoptosis in AC16 cells was detected by using flow cytometry (CytoFLEX, BECKMAN COULTER, USA). A total of 1 × 105 cells per group were harvested and stained by using Annexin V- fluorescein isothiocyanate (FITC)/propidium iodide (PI) cell Apoptosis Detection Kit (40302ES50, Yeasen Biotechnology, Shanghai, China). The cells were collected at 300 g and 4℃ for 5 min. The cells were washed with precooled PBS twice with 300 g and 4 ℃ for 5 min each time and collected. A total of 100 μL Binding Buffer (×1) was added to the suspension cells. The cells were gently mixed with 5 μL Annexin V-FITC and 10 μL of PI staining solution and reacted at RT and away from light for 15 min. The cells were added with 400 μL Binding Buffer (×1), mixed and placed on ice. The samples were detected by flow cytometry within 1 h. Data were analyzed by using FlowJo v10 (BD Bioscience, USA) to derive the apoptosis rate values for each sample.
Cell counting kit 8 (CCK-8)
The experimental procedure was conducted in accordance with the instructions provided by Solarbio (CA1210, Shanghai, China) for the CCK-8 assay, which is a widely utilized assay for assessing cell proliferation and cytotoxicity. In brief, AC16 cells were seeded at a density of 2000 cells per well in 96-well plates and incubated for 48 h. Subsequently, each well was supplemented with 10 μL of CCK-8 reagent. The cells were then incubated for 2 h before absorbance was measured at 450 nm.
2,3,5-Triphenyltetrazolium chloride (TTC) staining
Infarct size was determined by using TTC staining. Briefly, heart tissues were isolated and frozen before being sliced into sections. Subsequently, the samples were incubated at 37°C for 15 min in a solution of 1% TTC (17779, Sigma-Aldrich, China) before imaging.
TdT-mediated dUTP nick end labeling (TUNEL) staining assay
The apoptosis of cardiomyocytes was assessed by performing TUNEL staining on heart tissues. A TUNEL apoptosis assay kit (C1091, Beyotime, Shanghai, China) was employed by following previously described protocols. Subsequently, the stained heart tissue slices were covered with biotin labeling solution, incubated at 37℃ in the dark for 1 h, and washed three times with PBS. The samples were added with streptavidin-Horseradish Peroxidase (HRP) working solution, incubated for 30 min at RT, and then washed three times with PBS. The samples were added with 3,3’-diaminobenzidine (DAB) chromogen solution, incubated for 20 min at RT, and then washed three times with PBS. Nuclei were stained with hematoxylin (191202, Yulu Experimental Equipment Co., LTD, Nanchang, China) staining solution. The slices were then sealed for observation and photographed under a light microscope (BX-53, Olympus, Tokyo, Japan).
Statistical analysis
Statistical analyses were conducted by using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). The t-test was employed to analyze the differences between the two groups. Analysis of variance was used for multigroup comparative analysis. The Bonferroni post hoc test was then performed to assess the significance of differences between the two groups. The results are presented as the mean ± standard deviation, with statistical significance defined as P < 0.05. The figures were created by using PowerPoint [2021MSO, Microsoft® PowerPoint®, USA], Photoshop [13.0, Adobe® Photoshop® CS6, USA], and BioRender [©BioRender 2024, https://app.biorender.com/]).
RESULTS
MiR-3148 increased in AMI patients and mice, as well as AC16 cells
To elucidate the effect of miR-3148 on AMI, we assessed its expression levels in the serum of AMI patients undergoing PCI. The demographic and clinical characteristics of the AMI patients are summarized in Table 1. We observed that in patients with AMI, miR-3148 expression significantly elevated and then gradually declined following PCI treatment [Figure 1a]. Subsequently, we established the mouse models of AMI. The infarcted region, indicative of MI, was visualized as a white area through TTC staining [Figure 1b]. Using H&E staining, heart tissue analysis revealed significant interstitial edema within the myocardium, the partial coagulative necrosis of cardiomyocytes with a marked decrease in their number, and the infiltration of inflammatory cells in the model group [Figure 1c]. Compared with those in the Sham group, the myocardial cells in the model group showed a notable upregulation in miR-3148 expression [Figure 1d]. In vitro investigations were further conducted by utilizing AC16 cells subjected to OGD to mimic cellular conditions akin to AMI [Figure 1e]. Consistently, a significant augmentation in miR-3148 expression was observed [Figure 1f].
| AMI patients | Control (healthy individuals) | |||
|---|---|---|---|---|
| Age (years) | Gender | Diagnostics | Age (years) | Gender |
| 69 | Male | AMI (Antero-septal walls) | 53 | Male |
| 51 | Male | AMI (Anterior walls) | 71 | Male |
| 49 | Female | AMI (Inferior walls) | 50 | Male |
| 73 | Male | AMI (Anterolateral walls) | 73 | Male |
| 39 | Female | AMI (Anterior walls) | 73 | Male |
| 55 | Male | AMI (Anterior walls) | 67 | Female |
| 58 | Male | AMI (Posterior walls) | 52 | Female |
| 63 | Male | AMI (Lateral walls) | 52 | Female |
| 59 | Female | AMI (Posterior walls) | 71 | Female |
| 67 | Male | AMI (Anterior walls) | 71 | Female |
Both gender and age are not statistically significant (P>0.05). AMI: Acute myocardial infarction
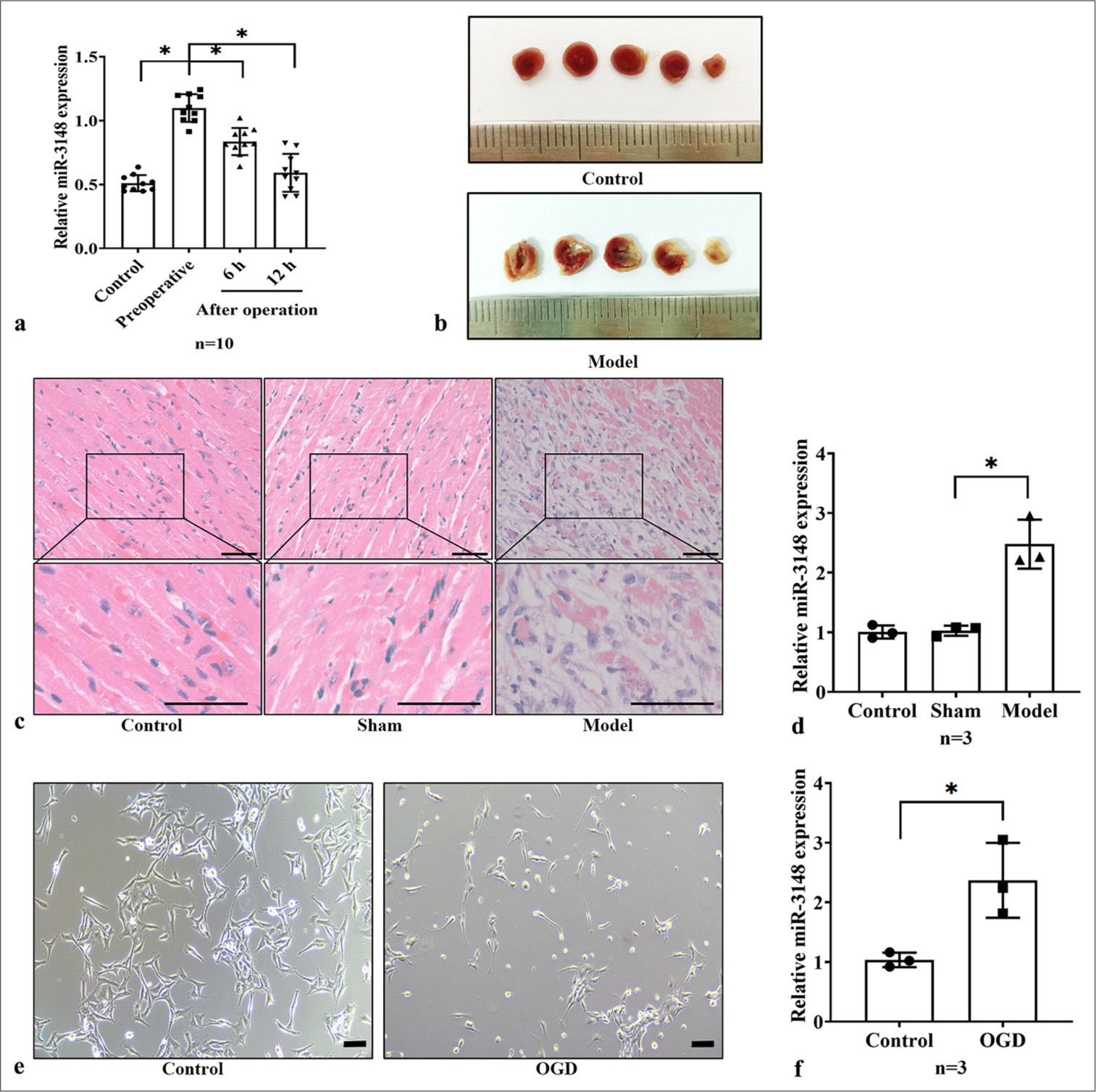
- miR-3148 increased in AMI models. (a) The expression level of miR-3148 in patients with AMI was evaluated by using qRT-PCR. n = 10 in each group. (b) TTC staining of mice myocardium in the control and model groups. (c) H&E staining of cardiac tissues from Control, Sham and Model mice. Bar = 50 μm. (d) The quantification of miR-3148 expression was conducted by using qRT-PCR in the Control, Sham and AMI mice, n = 3 in each group. (e) Cells morphology were observed under control condition and after OGD. Bar = 100 μm. (f) miR-3148 expression levels in the Control and OGD AC16 cells were measured by qRT-PCR, n = 3 in each group. All analyses, ✶: P < 0.05. AMI: Acute myocardial infarction, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, OGD: Oxygen and glucose deprivation, TTC: 2,3,5-Triphenyltetrazolium chloride, H&E: Hematoxylin and eosin, mRNA: Messenger RNA.
Inhibition of miR-3148 could promote myocardial mitophagy and reduce apoptosis in AMI mice
To investigate the in vivo implications of miR-3148, we administered miR-3148 antagomir via tail vein injection to assess its impact on mitophagy and apoptosis in mouse myocardial cells. Following administration, there was a notable increase in the protein levels of PINK1 and Parkin, Beclin1, and LC3 II/I accompanied by a significant decrease in p62, which are indicative of key hallmarks of the classical mitophagy pathway [Figure 2a and b]. Concurrently, the mRNA expression levels of PINK1 and Parkin were upregulated [Figure 2c], further corroborating the activation of mitophagy. Moreover, compared to the AMI Model group, the protein levels of apoptotic markers, including Caspase 3, Caspase 9, and Cyt C, exhibited a significant decrease in the miR-3148 antagomir-treated group [Figure 2d and e]. This attenuation of apoptosis was also validated by TUNEL staining, wherein the myocardial apoptosis level was markedly reduced in the miR-3148 antagomir group compared to the AMI Model group [Figure 2f and g].
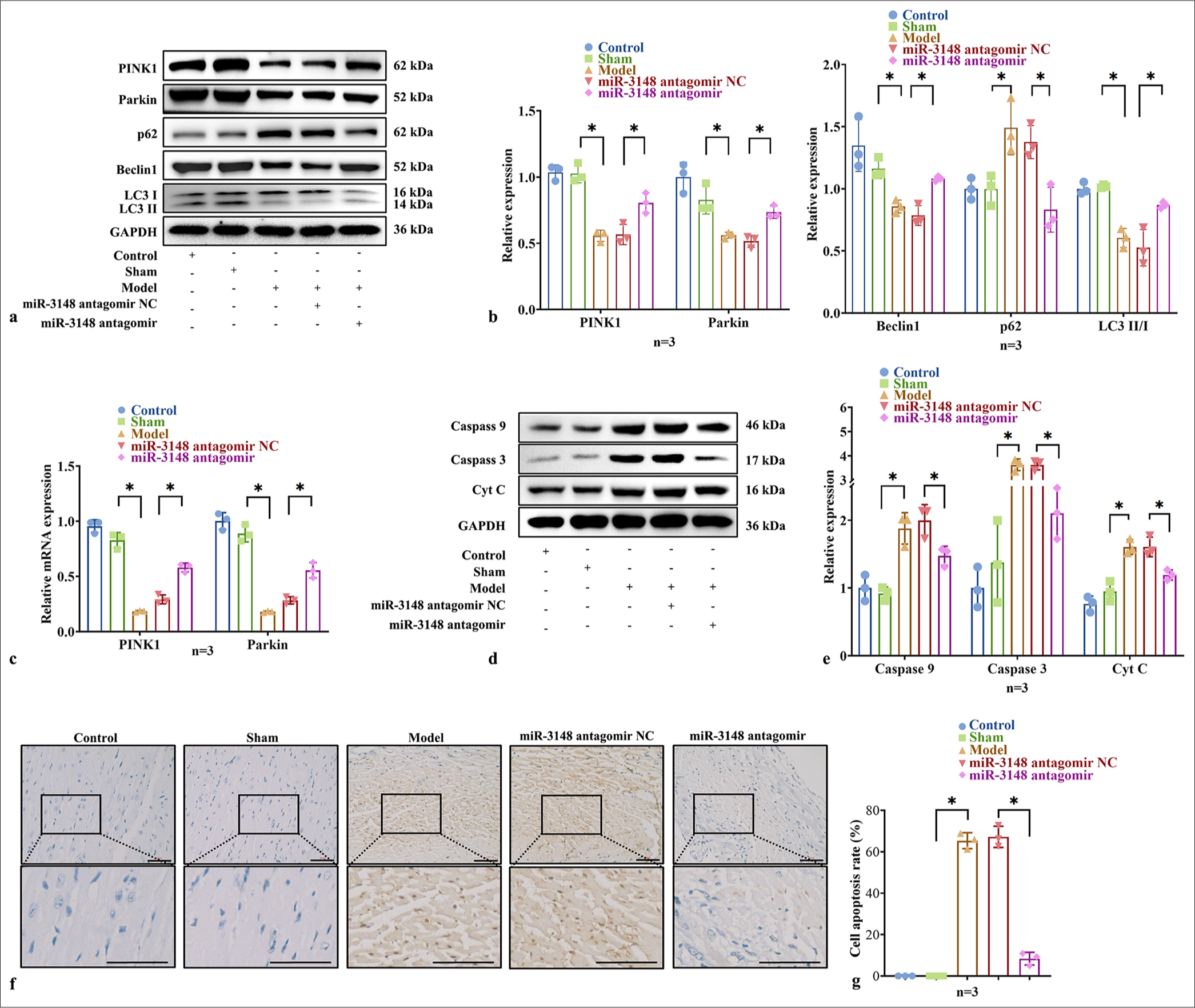
- Inhibition of miR-3148 could promote myocardial mitophagy and reduce apoptosis in AMI mice. (a and b) WB analysis was performed to examine the expression levels of PINK1, Parkin, Beclin1, p62, LC3 I and LC3 II proteins associated with mitophagy. (c) The PINK1 and Parkin expression were measured by qRT-PCR. (d and e) Levels of apoptosis-related proteins, including Caspase 9, Caspase 3, and Cyt C, were evaluated through WB analysis. (f) TUNEL staining of mice myocardium. Apoptotic cells that were TUNEL-positive appear brown, with hematoxylin staining revealing blue nuclei. Cells that were TUNEL-positive typically cannot be further stained with hematoxylin. Bar = 50 μm. (g) Comparison of the number of TUNEL positive cells of mice among groups. All analyses, n = 3 in each group; ✶: P < 0.05. WB: Western blot, AMI: Acute myocardial infarction, PINK1: PTEN-induced kinase 1, LC3: Microtubule-associated proteins 1A/1B light chain 3B, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, Caspase: Cysteine-aspartic acid protease, Cyt C: Cytochrome C, TUNEL: TdT-mediated dUTP nick end labeling, Parkin: Parkin RBR E3 ubiquitin protein ligase, p62: Sequestosome 1, mRNA: Messenger RNA.
Suppression of miR-3148 affected the viability of OGD cardiomyocytes and promoted mitophagy
To investigate the role of miR-3148 in cardiomyocyte viability, we examined AC16 cell activity and apoptosis under OGD conditions. The CCK-8 experiment clearly demonstrated a decrease in cell viability in the OGD group, while higher cell viability in the miR-3148 inhibitor group suggested partial alleviation of this inhibitory effect. However, compared to the control group, the cell viability remained lower in the miR-3148 inhibitor group, indicating incomplete restoration of normal cell proliferation ability by the inhibitor [Figure 3a]. The significantly increased rate of apoptosis in the OGD group indicates that OGD treatment exerted a pronounced apoptotic effect on AC16 cells. Conversely, decreased apoptosis rates were observed with suppressed miR-3148 expression, suggesting an inhibitory effect of the inhibitor on cell apoptosis [Figure 3b and c]. Compared with OGD cells, significant decreases were observed in protein levels of Caspase 3, Caspase 9, and Cyt C within the miR-3148 inhibitor group [Figure 3d and e]. Suppression of miR-3148 resulted in increased protein levels of PINK1, Parkin, Beclin1, LC3 II/I, and decreased levels of p62 [Figure 3f and g]. Meanwhile, mRNA expressions of PINK1 and Parkin were downregulated [Figure 3h]. These results suggest that miR-3148 regulates mitophagy and apoptosis during myocardial ischemia injury.
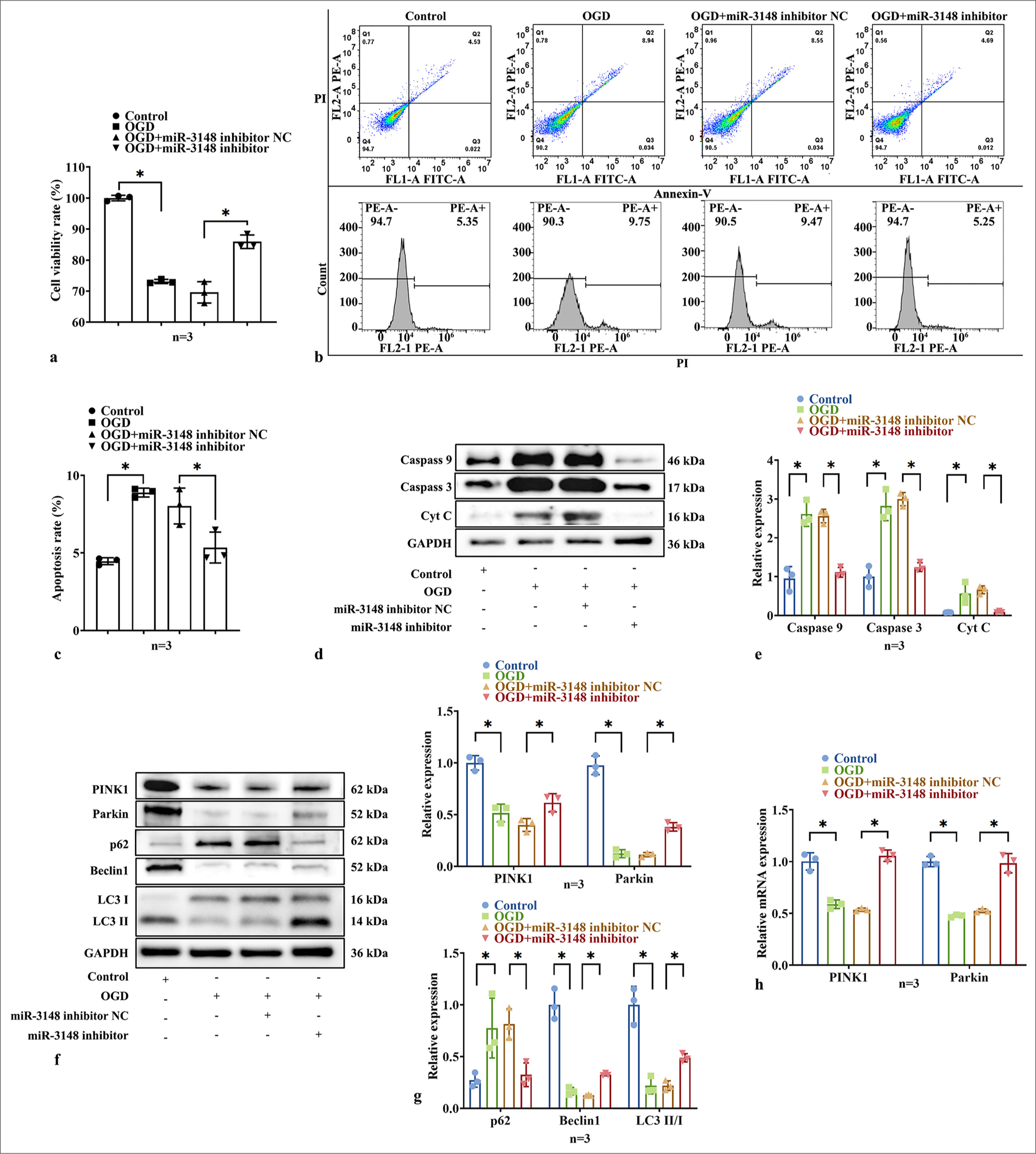
- Suppression of miR-3148 affected the viability of OGD cardiomyocytes and promoted mitophagy. (a) CCK8 detection of cells activity. (b and c) The assessment of apoptosis in AC16 cells was conducted through the utilization of Flow Cytometry. (d and e) The levels of apoptosis-related proteins, including Caspase 9, Caspase 3, and Cyt C, were evaluated through WB analysis. (f and g) WB analysis was performed to examine the expression levels of PINK1, Parkin, Beclin1, p62, LC3 I and LC3 II proteins associated with mitophagy. (h) The PINK1 and Parkin expression were measured by qRT-PCR. All analyses, n = 3; ✶: P < 0.05. PINK1: PTEN-induced kinase 1, Caspase: Cysteine-aspartic acid protease, Cyt C: Cytochrome C, OGD: Oxygen and glucose deprivation, LC3: Microtubule-associated proteins 1A/1B light chain 3B, WB: Western blot, Parkin: Parkin RBR E3 ubiquitin protein ligase, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, p62: Sequestosome 1, mRNA: Messenger RNA.
KLF6 is the target of miR-3148 and is downregulated in AMI mice and OGD cardiomyocytes
We confirmed that the upregulation of miR-3148 exacerbates apoptosis in myocardial ischemia injury by inhibiting mitophagy. In order to elucidate the mechanism of miR-3148, we investigated its target genes related to mitophagy. There were 291 predicted targets for hsa-miR-3148 in microRNA Target Prediction Database (miRDB), with Target Score ≥95. In miDIP, 172 predicted targets for hsa-miR-3148 were classified as “Very High” in score. TargetScan predicted 300 targets for hsa-miR-3148 with a context++ score ≥−0.4. A total of 36 target genes were common across “microRNA Data Integration Portal,” “miRDB,” and “TargetScan,” as shown in the Venn diagram (https://bioinfogp.cnb.csic.es/tools/venny/index.html). We found that KLF6 is one of candidate targets of miR-3148 [Figure 4a]. Figure 4b illustrates the putative binding sites between miR-3148 and the 3’-UTR of KLF6 in www.targetscan.org. To investigate whether miR-3148 directly targets KLF6 through its 3’UTR and regulates its expression, we designed GP-miRGLOKLF6-3’-UTR-WT and GP-miRGLO-KLF6-3’-UTR-MUT luciferase reporter plasmids. The luciferase reporter activities were reduced by transfection with miR-3148 mimics in the WT group but not in the MUT group, indicating a direct interaction between miR-3148 and KLF6 [Figure 4c]. We then performed an in vivo experiment, which demonstrated a decrease in cardiac KLF6 protein levels in AMI mice, and an increase in myocardial KLF6 expression following the downregulation of miR-3148 [Figure 4d and e]. Meanwhile, we transfected the AC16 cells with either the miR-3148 mimic or miR-3148 inhibitor [Figure 4f]. Upregulation of miR-3148 inhibited KLF6 expression [Figure 4g and h], while its downregulation promoted KLF6 expression [Figure 4i and j]. It indicated the regulating correlation between miR-3148 and KLF6.
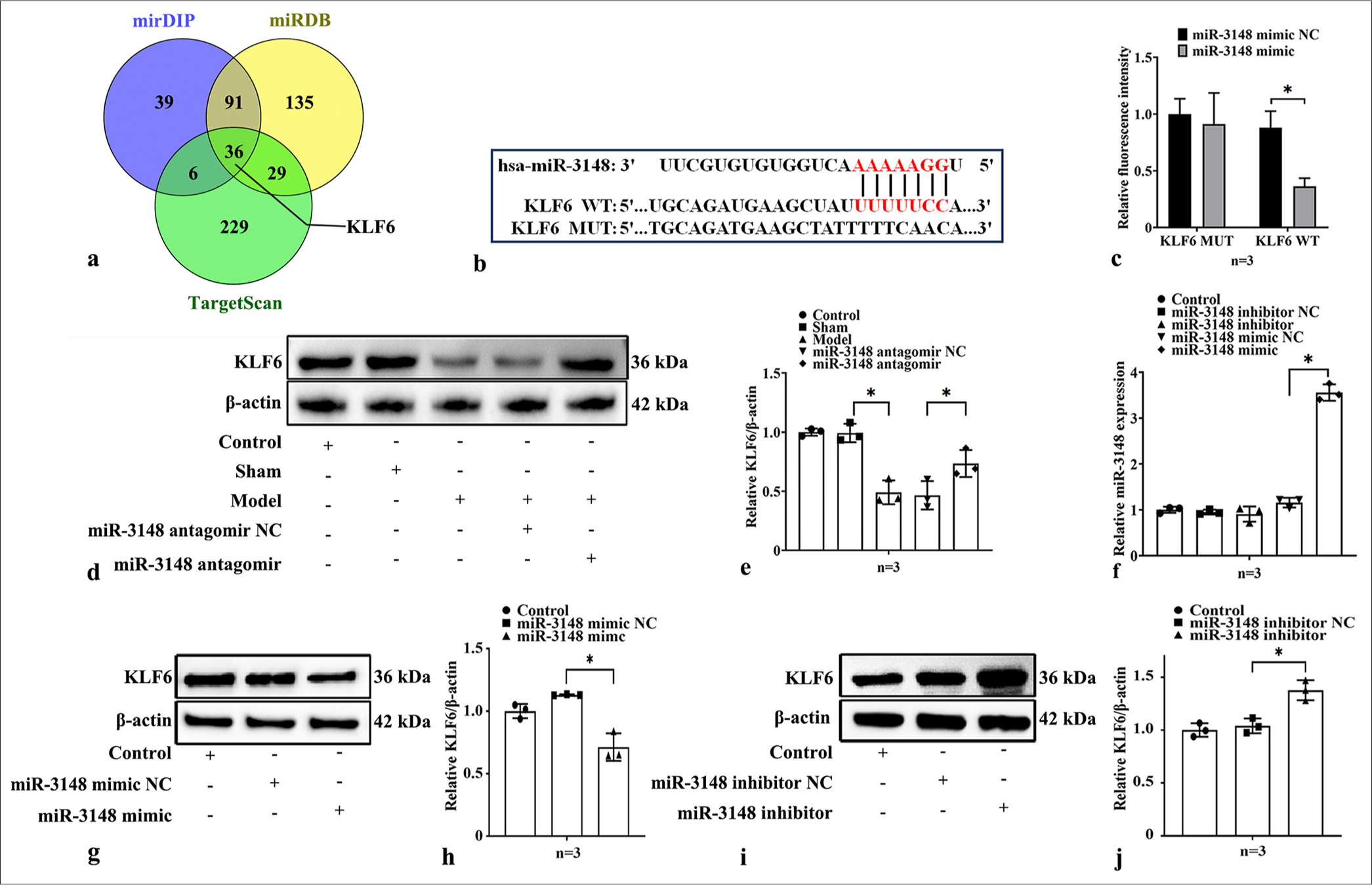
- KLF6 is the target of miR-3148 and is down-regulated in AMI mice and OGD cardiomyocytes. (a) Hsa-miR-3148 targets were predicted in the miRDB, mirDIP and TargetScan databases. (b) The illustration of predicted 3'-UTR binding sites for KLF6 of miR-3148 in www.targetscan.org. (c) Luminescence levels were measured in cells transfected with luciferase reporter plasmids harboring either the WT (KLF6 WT) or MUT (KLF6 MUT) miR-3148 binding sites within the 3’-UTR of KLF6, comparing both the Control group and the group treated with miR-3148 mimic. (d and e) The expression levels of KLF6 were evaluated using WB analysis in myocardial tissues of mice. (f) The expression level of miR-3148 was measured by qRT-PCR after transfecting AC16 with miR-3148 mimic or miR-3148 inhibitor. (g and h) Up-regulation of miR-3148 inhibited KLF6 expression, (i and j) while its down-regulation promoted KLF6 expression. All analyses, n = 3; ✶: P < 0.05. KLF6: Krüppel-like factor 6, AMI: Acute myocardial infarction, OGD: Oxygen and glucose deprivation, miRDB: MicroRNA target prediction database, mirDIP: MicroRNA data integration portal, 3’-UTR: 3’-untranslated region, WT: Wild type, MUT: Mutant, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, WB: Western blot.
Suppression of KLF6 promoted the apoptosis of AC16 cells under OGD conditions and reduced mitophagy
Based on previous results, we suggested that miR-3148 regulates mitophagy and apoptosis by targeting KLF6. To explore the assumption, we downregulated KLF6 expression in AC16 cells [Figure 5a], then detected mitophagy and apoptosis by qPCR, CCK-8, flow cytometry, and WB. The CCK-8 experiment demonstrated that under OGD conditions, the cell viability in the si-KLF6 group exhibited a significantly lower level compared to that in the si-KLF6-NC group [Figure 5b], indicating that downregulation of KLF6 expression exerted a more pronounced inhibitory effect on cell proliferation. The si-KLF6 group exhibited a significantly increased apoptosis rate under OGD conditions, as compared to the si-KLF6-NC group [Figure 5c and d], indicating that downregulation of KLF6 expression could enhance cellular apoptosis. To investigate the impact of KLF6 on mitophagy and apoptosis, WB analysis was conducted to evaluate the expression levels of proteins associated with PINK1/Parkin-mediated mitophagy and apoptosis. The protein of Caspase 3, Caspase 9, and Cyt C was increased significantly in si-KLF6 group [Figure 5e and f]. By downregulated KLF6, the protein of PINK1, Parkin, Beclin1, LC3 II/I were decreased, and p62 were increased significantly [Figure 5g and h]. Meanwhile, the mRNA expressions of PINK1 and Parkin were downregulated [Figure 5i]. It was evident that knock-down of KLF6 could decrease mitophagy and further increase apoptosis in myocardial ischemia injury.
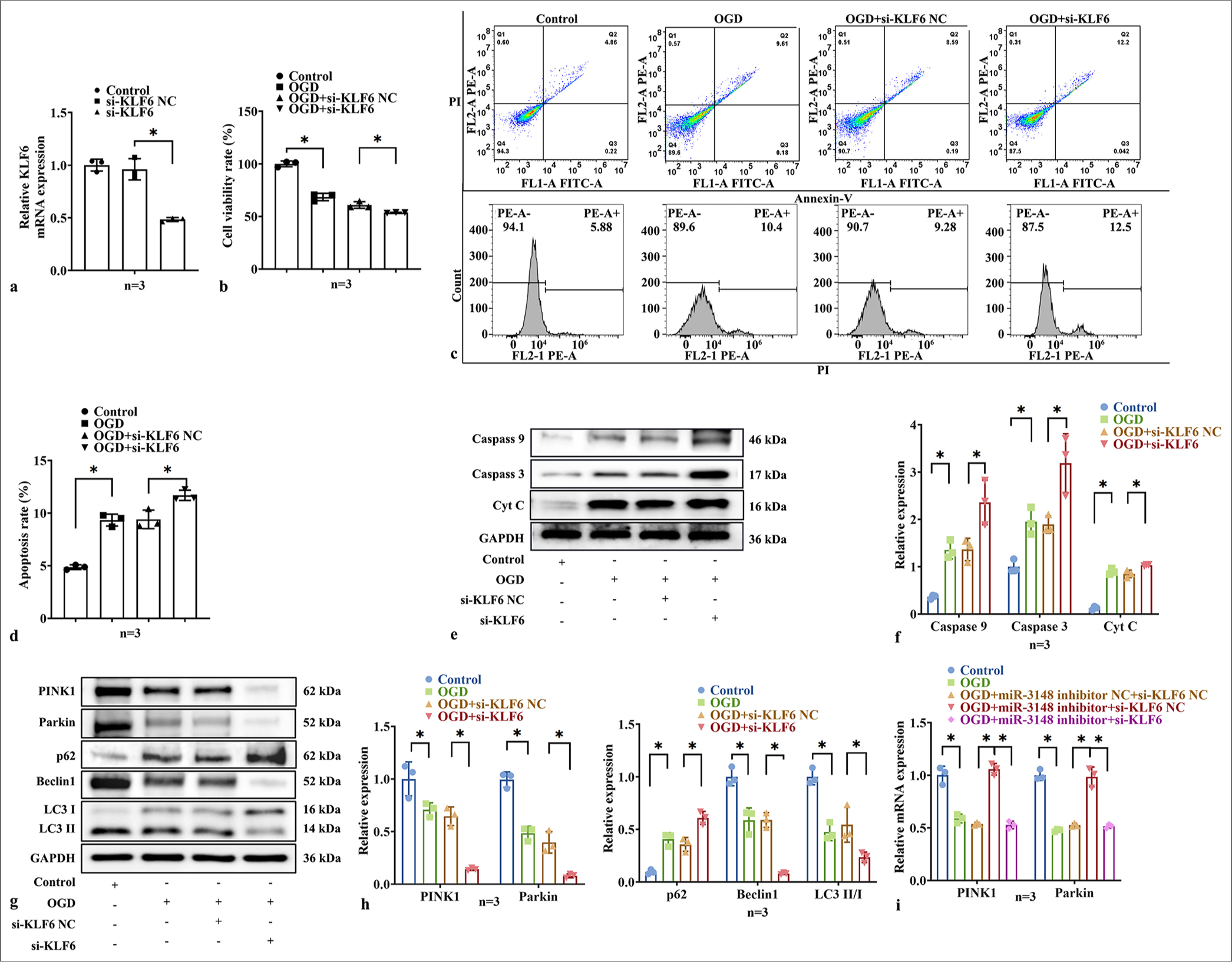
- Suppression of KLF6 promoted the apoptosis of AC16 cells under OGD conditions and reduced mitophagy. (a) The expression level of KLF6 was measured by qRT-PCR after transfecting AC16 cells with si-KLF6. (b) CCK8 detection of cells activity. (c and d) The measurement of apoptosis in AC16 cells was conducted using Flow Cytometry. (e and f) After treatment with si-KLF6, the levels of apoptosis-related proteins, including Caspase 9, Caspase 3, and Cyt C, (g and h) and the levels of mitophagy-related proteins of PINK1, Parkin, Beclin1, p62, LC3 I and LC3 II, were evaluated through WB analysis. (i) The mRNA expression levels of PINK1 and Parkin were detected by qRT-PCR. All analyses, n = 3; ✶: P < 0.05. KLF6: Krüppel-like factor 6, Caspase: Cysteine-aspartic acid protease, Cyt C: Cytochrome C, Parkin: Parkin RBR E3 ubiquitin protein ligase, CCK-8: Cell counting kit 8, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, OGD: Oxygen and glucose deprivation, si-KLF6: Small interfering RNA targeting KLF6, PINK1: PTEN-induced kinase 1, LC3: Microtubule-associated proteins 1A/1B light chain 3B, WB: Western blot, p62: Sequestosome 1, mRNA: Messenger RNA.
MiR-3148 regulated mitophagy and apoptosis through KLF6
We confirmed the regulatory roles of both miR-3148 and KLF6 in controlling mitophagy and apoptosis, with miR-3148 specifically targeting the KLF6 gene. In addition, we aimed to investigate the potential effect of miR-3148 on KLF6. Therefore, we downregulated KLF6 in the miR-3148 inhibitor cells under the OGD conditions. We performed CCK-8 assay and flow cytometry to observe the relative cell activity and apoptosis of AC16 cells. We found a significant increase in cell activity and a decrease in apoptosis in the miR-3148 inhibitor group. However, the downregulation of KLF6 in the miR-3148 inhibitor group could reverse the effect [Figure 6a-c]. Then, the proteins of mitophagy and apoptosis were detected by WB. The levels of apoptosis-associated proteins decreased in the miR-3148 inhibitor group; however, the downregulated KLF6 in the miR-3148 inhibitor group could reverse the effect under the OGD conditions [Figure 6d and e]. Moreover, the inhibition of miR-3148 resulted in a notable elevation in the mRNA levels of PINK1 and Parkin expressions. The downregulation of KLF6 was able to counteract the effects induced by inhibiting miR-3148 under OGD conditions [Figure 6f]. By downregulated KLF6 in the miR-3148 inhibitor cells, the protein of PINK1, Parkin, Beclin1, and LC3 II/I was decreased, and p62 was increased significantly under OGD conditions, as compared to the miR-3148 inhibitor+si-KLF6-NC group [Figure 6g and h]. Hence, these findings imply that KLF6 plays a crucial role in the regulation of mitophagy through PINK1/Parkin-mediated pathways, as well as in apoptosis during myocardial ischemic injury.
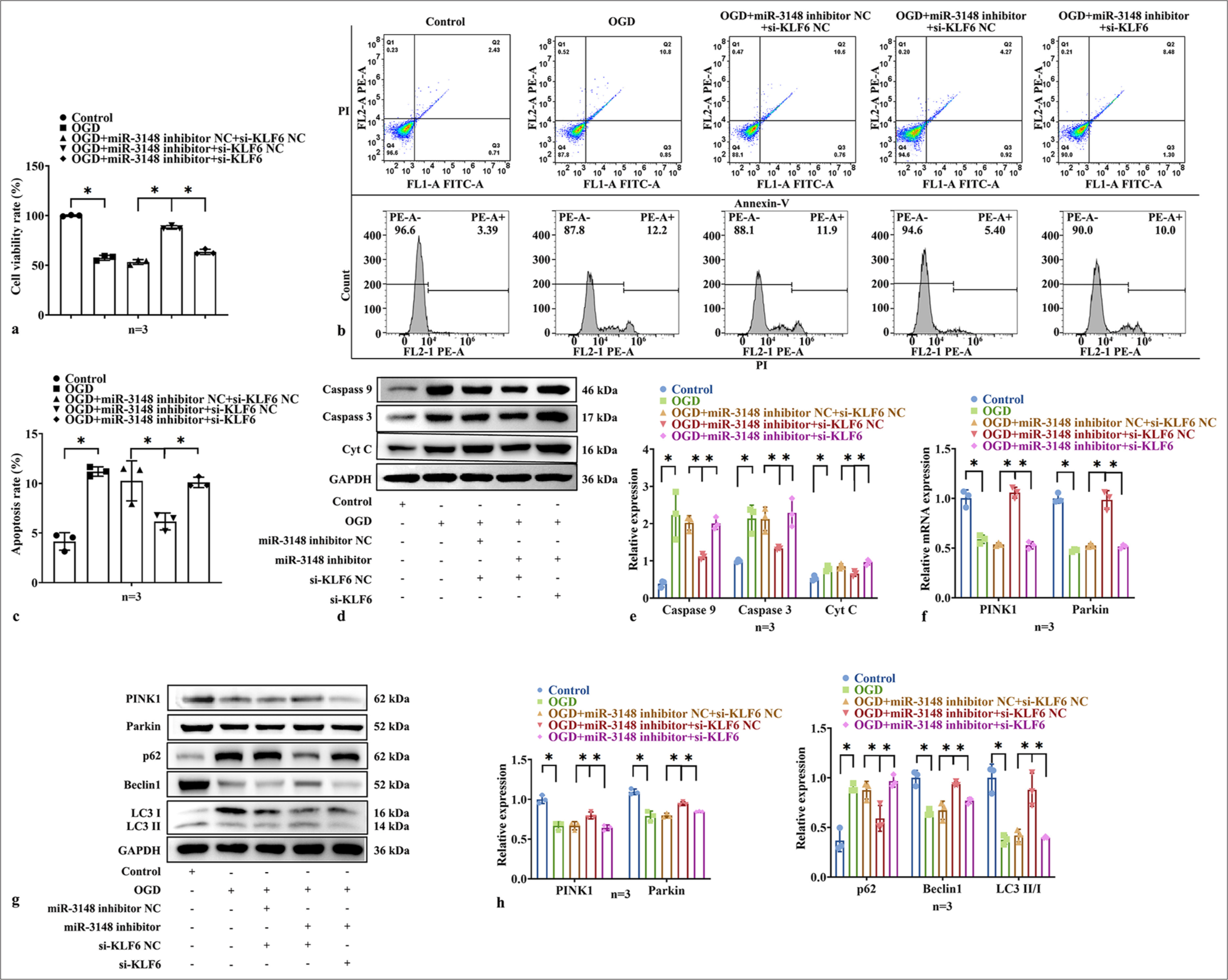
- miR-3148 regulated mitophagy and apoptosis though KLF6. (a) CCK-8 detection of cells activity. (b and c) The measurement of apoptosis in AC16 cells was conducted using flow cytometry. (d and e) The levels of apoptosis-related proteins of Caspase 9, Caspase 3, and Cyt C were evaluated through WB analysis. (f) The mRNA expression levels of PINK1 and Parkin were detected by qRT-PCR. All analyses, n = 3; ✶: P < 0.05. (g and h) The levels of mitophagy-related proteins of PINK1, Parkin, Beclin1, p62, LC3 I and LC3 II, were evaluated through WB analysis. Cyt C: Cytochrome C, Parkin: Parkin RBR E3 ubiquitin protein ligase, CCK-8: Cell counting kit 8, LC3: Microtubule-associated proteins 1A/1B light chain 3B, WB: Western blot, Caspase: Cysteine-aspartic acid protease, PINK1: PTEN-induced kinase 1, KLF6: Krüppel-like factor 6, qRT-PCR: Quantitative reverse transcription polymerase chain reaction, p62: Sequestosome 1, mRNA: Messenger RNA.
DISCUSSION
In recent years, numerous studies have reported the involvement of miRNAs in the mitophagy and apoptosis of cardiomyocytes following MI.[19] Among these miRNAs, miR-3148 has been identified as a key player in various human diseases. For instance, microRNA chip analysis revealed that miR-3148 may function as an onco-miRNA candidate. Meanwhile, the stable expression of miR-3148 was found to confer growth advantage to human colon cancer cells.[20] Functional experiments conducted in vitro demonstrated that miR-3148 negatively regulates otopetrin 2 (OTOP2) expression in colorectal cancer cells, and silencing OTOP2 reduces Caspase 3/9 activity while promoting cell migration, proliferation and epithelial mesenchymal transformation.[10] In cardiovascular disease research, Cakmak et al. used microarray methods to analyze the serum levels of 20 patients with stable cardiac function (New York Heart Association [NYHA] II), 22 patients with decompensated chronic congestive heart failure (CHF) (NYHA III and IV), and 15 healthy controls. Their results showed that the serum levels of miR-3148 were significantly higher in patients with CHF than in those without and revealed a significant correlation between serum high-sensitivity C-reactive protein/N-terminal pro b-type natriuretic peptide (NT-proBNP) levels and circulating levels of this particular miRNA.[21] We observed that a significant upregulation of miR-3148 in the serum of AMI patients, which was subsequently downregulated upon removal of myocardial ischemia. Further investigations revealed the elevated expression levels of miR-3148 in the hearts of mice with AMI and AC16 cell models under OGD conditions. Cardiomyocyte apoptosis is a critical contributor to ventricular dysfunction and remodeling, necessitating an exploration of its underlying molecular mechanisms for potential therapeutic targets in AMI treatment. Throughout apoptosis, Caspase 9 functions as an initiator protein, while Caspase 3 operates as an executor protein. When impaired, mitochondria release Cyt C into the cytoplasmic milieu, where it interacts with apoptosis protease-activating factor-1 (Apaf-1), forming a polymeric complex termed the apoptosome. The amino-terminal segment of apoptotic bodies contains Caspase activation and recruitment domain (CARD) sequences, which selectively engage with the CARD sequence of Caspase 9, thereby initiating the downstream activation of Caspase 3.[22] Upon receiving apoptotic signals, Bcl-2 associated X protein (Bax) promotes Cyt C release from mitochondrial membranes; however, excessive B-cell lymphoma-2 (Bcl-2) inhibits Cyt C release by binding to Bax and Apaf-1, thus preventing procaspase activation.[23] In this study, inhibition of miR-3148 expression resulted in decreased expressions of Caspase 9, Caspase 3, and Cyt C in both in vivo and in vitro experiments. These findings suggest that the effective prevention of OGD-induced apoptosis of AC16 cells and reduction of cardiomyocyte apoptosis in mice with AMI can be achieved by inhibiting miR-3148. Therefore, our results indicate a significant role for miR-3148 in inducing apoptosis during the progression of AMI.
Mitochondria play a pivotal role as the primary trigger for cardiomyocyte injury, as over 90% of the energy supply for cardiomyocytes originates from these organelles. The severity of mitochondrial apoptosis and damage is directly correlated with the extent of cardiomyocyte death.[24] Improving the function of mitochondria in heart muscle cells is a vital objective for therapeutic intervention aimed at delaying the development and advancement of cardiovascular ailments.[1,25] The current research has revealed that mitophagy operates at molecular, organelle, and cellular levels to ensure cell homeostasis, preserve normal cellular structure and function, and facilitate proper mitochondrial functioning.[26] Disrupted mitophagy can lead to dysfunction or even demise of cardiomyocytes; however, promoting mitophagy can mitigate tissue cell damage caused by abnormal mitochondria while restoring mitochondrial energy metabolism.[27] Among the signaling pathways involved in mitophagy, the PINK1/Parkin pathway is considered a classical one due to its ability to detect the formation and degradation of autophagosomes within the mitochondria. When mitochondria undergo damage and experience a decrease in the membrane potential, the activity of proteolytic enzymes is inhibited, leading to the phosphorylation of PINK1 and its subsequent accumulation on the damaged outer mitochondrial membrane.[28] Parkin functions as an E3 ubiquitin ligase that can be recruited by PINK1 on the outer mitochondrial membrane to catalyze ubiquitination of relevant proteins present on the surface of this organelle. Ubiquitinated mitochondria are recognized by autophagy-related protein p62, facilitating their binding with homologous proteins such as LC3 II, located on lysosomal membranes for the sequestration of defective mitochondria. This process induces selective removal through autophagy, enabling MQC mechanisms aimed at limiting damage expansion and inhibiting apoptosis.[29] Simultaneously, autophagy-related proteins continue to facilitate the conversion of cytoplasmic LC3 I into membrane-bound LC3 II, and the ratio of LC3 II/I is commonly employed as an indicator of cellular autophagic activity.[30] Beclin1 serves as a pivotal regulator in autophagy. Numerous autophagy regulatory proteins modulate the levels of autophagy by interacting with distinct domains of Beclin1 to form protein complexes. The anti-apoptotic protein Bcl-2 can also bind to Beclin1 through its BH3 domain, thereby inhibiting Beclin1-dependent autophagy and promoting apoptosis. Consequently, through interactions with either autophagy-associated or apoptosis-associated proteins, Beclin1 determines whether cells undergo autophagy or apoptosis.[31] We aimed to investigate the impact of miR-3148 expression changes on myocardial mitochondrial function in the heart. Our findings from both in vivo and in vitro model experiments revealed that downregulation of miR-3148 led to increased expressions of PINK1 and Parkin, as well as elevated levels of Beclin1, and LC3 II/I ratios. In addition, p62 expression decreased, indicating a restoration of mitophagy function. The decline in mitophagy induced the enhanced apoptosis of cardiomyocytes, thus highlighting the importance of miR-3148 as a key influencing factor.
To investigate the potential mechanism underlying mitophagy, we conducted target gene prediction for miR-3148. KLF6 was selected as the study object due to its highest score and significant role in protecting mitochondrial metabolism. Previous study has demonstrated the binding of KLF6 to transcriptional binding sites located on the promoter region of synthesis of Cytochrome C oxidase 2 (SCO2), a critical gene involved in preventing Cyt C release and activating endogenous apoptotic pathways, thereby regulating linear subfunction and mitophagy.[13] Luciferase experiments confirmed the association between miR-3148 and KLF6, with up-regulation of miR-3148 leading to decreased expression of KLF6, while down-regulation resulted in increased expression. Furthermore, inhibiting the expression of KLF6 could reverse the promotion of mitophagy caused by downregulated miR-3148.
KLF6 plays a crucial regulatory role in cell survival and apoptosis, as well as in the regulation of macrophage polarization and initiation of pro-inflammatory response. A recent study has demonstrated that KLF6 could modulate the expression of hypoxia-inducing factors in macrophages, thereby regulating both hypoxic conditions and inflammatory responses.[32] Horne et al. observed that loss of KLF6 in podocytes led to reduced SCO2, resulting in increased mitochondrial damage and activation of the intrinsic apoptotic pathway under diabetic conditions. Conversely, overexpression of KLF6 significantly mitigated mitochondrial damage and apoptosis in cultured human podocytes exposed to hyperglycemic conditions.[33] KLF6 exhibits complex biological functions in the myocardium, exerting both detrimental effects that exacerbate myocardial injury and certain protective roles. However, the specific mechanisms underlying these effects remain controversial. Qiu et al. demonstrated that KLF6 exacerbates myocardial I/R by activating the ferroptosis pathway mediated by fatty acyl-CoA synthetase 4.[34] In contrast, Fang et al. suggested that KLF6 plays a detrimental role in hypoxia/reoxygenation-induced human cardiomyocyte injury by regulating m6A RNA methylation modification pathways.[35] However, the role of KLF6 in the myocardium is multifaceted. We observed a decrease in KLF6 expression in the myocardial cells of mice with AMI and OGD-treated AC16 cells. In OGD-treated AC16 cells, downregulation of KLF6 resulted in the decreased expressions of PINK1 and Parkin, as well as Beclin1 and LC3 II/I, while p62 expression was increased, indicating inhibition of mitophagy. The proteins of apoptosis of Caspase 3, Caspase 9, and Cyt C were increased, indicating the promotion of apoptosis.
The present study has certain limitations that need to be acknowledged. First, the relatively small sample size may limit the generalizability and statistical power of its results. This constraint may affect the robustness of its conclusions and warrants caution when extrapolating its findings to larger populations. Second, our investigation was limited to in vitro experiments focusing on the regulation of PINK1/Parkin-mediated mitophagy and its exacerbation of mitochondrial dysfunction through the targeted inhibition of KLF6 expression by miR-3148. However, further research is warranted to validate these findings in an in vivo setting. Such research is currently being pursued as part of our ongoing project. Third, the signaling pathways involving KLF6/PINK1/Parkin and their interaction network with respect to mitophagy require additional elucidation for a comprehensive understanding of cardiomyocyte dysfunction. Finally, additional research is required to ascertain the clinical importance of miR-3148 in myocardial ischemia despite our comprehensive examination of its effects on mitophagy associated with myocardial ischemia and its underlying mechanisms.
SUMMARY
In conclusion, our results indicate that the regulation of PINK1/Parkin-mediated mitophagy in cardiomyocytes is influenced by miR-3148. MiR-3148 is implicated in myocardial ischemia through the downregulation of KLF6 and inhibition of PINK1/Parkin-associated mitophagy. At the same time, the increased expression levels of Cyt C, Caspase 3, and Caspase 9 aggravated apoptosis. The modulation of the mitophagy pathway involving miR-3148 and the KLF6/PINK1/Parkin axis may represent a promising target for novel drug development aimed at treating myocardial ischemia, thereby offering a new therapeutic strategy for acute coronary syndromes [Figure 7].
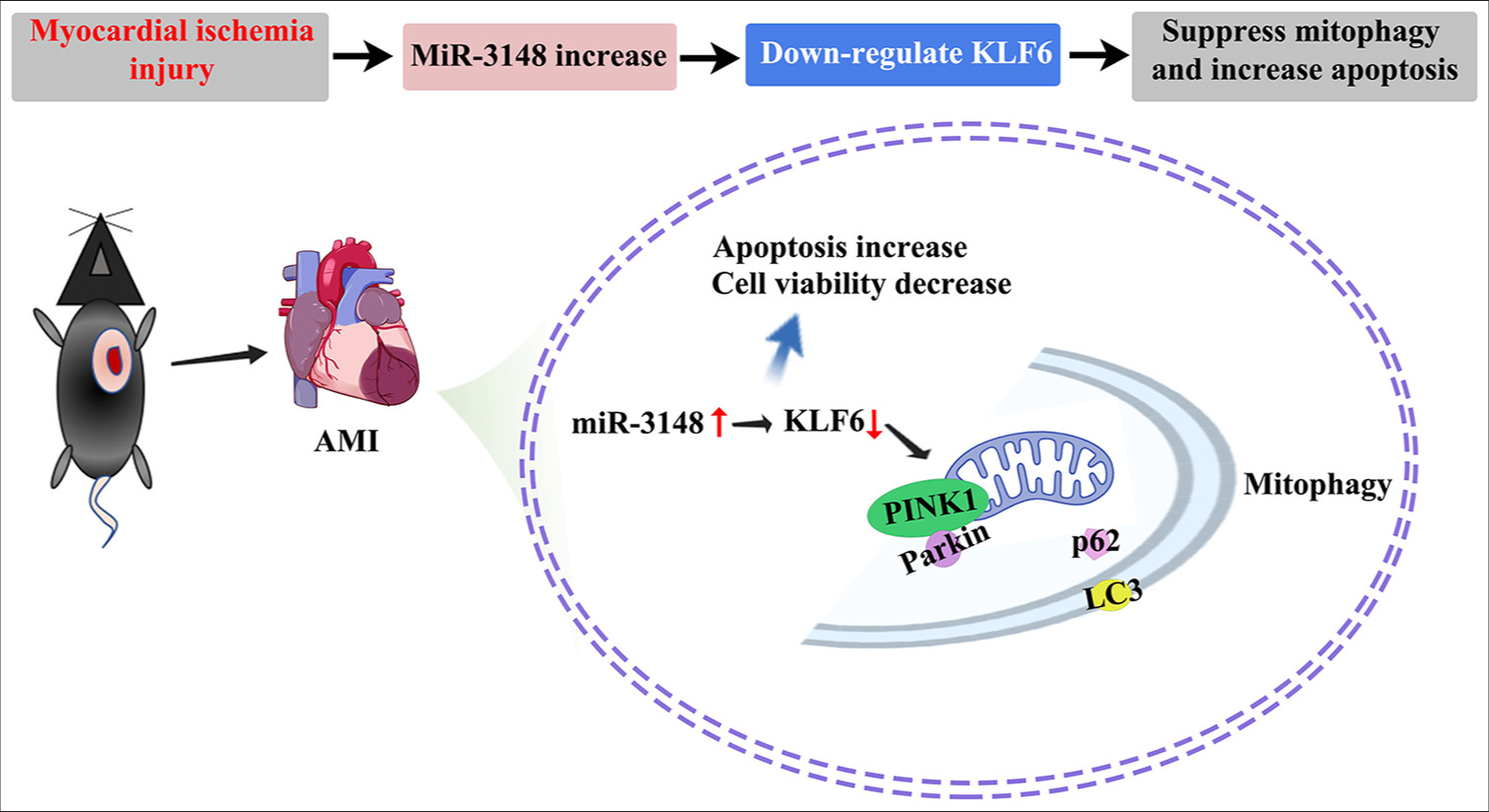
- The mechanism of miR-3148 attenuated PINK1/Parkin-mediated mitophagy and aggravated apoptosis in myocardial ischemia injury. In cases of myocardial ischemia injury, there is an up-regulation of miR-3148 which exacerbates apoptosis and hinders mitophagy. MiR-3148 plays a crucial role in PINK1/Parkin-mediated mitophagy by suppressing the mitochondria-related gene KLF6. Moreover, the decrease in KLF6 expression may impede the fusion between autophagosomes and lysosomes by reducing PINK1 accumulation on the outer membrane of mitochondria, resulting in reduced recruitment of Parkin. Consequently, this condition leads to an accumulation of p62, receptor proteins located on the outer membrane of mitochondria, causing a decline in Parkin ubiquitin products and impaired binding with LC3 for autophagy maturation. Therefore, inhibition of KLF6-mediated mitophagy contributes to the buildup of damaged mitochondria and promotes cellular apoptosis. Parkin: Parkin RBR E3 ubiquitin protein ligase, KLF6: Krüppel-like factor 6, PINK1: PTEN-induced kinase 1, p62: Sequestosome 1, LC3: Microtubule-associated proteins 1A/1B light chain 3B.
DATA AVAILABILITY STATEMENTS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
ABBREVIATIONS
AMI: Acute myocardial infarction
Cyt C: Cytochrome C
H&E: Hematoxylin and eosin
H/R: Hypoxia/reoxygenation
I/R: Ischemia-reperfusion injury
KLF6: Kruppel-like factor 6
LC3: Microtubule-associated proteins 1A/1B light chain 3B
MQC: Mitochondrial quality control system
mtDNA: Mitochondrial DNA
MI: Myocardial infarction
OGD: Oxygen and glucose deprivation
p62: Sequestosome 1
PARK2/Parkin: Parkin RBR E3 ubiquitin-protein ligase
PCI: Percutaneous coronary intervention
PINK1: PTEN-induced kinase 1
ROS: Reactive oxygen species
RT: Room temperature
TTC: 2,3,5-Triphenyltetrazolium chloride
ACKNOWLEDGMENT
This study thanks the support of Guangxi Key Clinical Department Construction Project.
AUTHOR CONTRIBUTIONS
CSH and LX: Designed the study; all authors conducted the study; YH and CSH: Collected and analyzed the data. YH and QJW: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, took public responsibility for appropriate portions of the content, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work were appropriately investigated and resolved.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All experiments were approved by the Ethics Committee of The Second Affiliated Hospital of Guangxi Medical University (2023-KY <0918>), and conducted in accordance with international ethical guidelines, the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the declaration of Helsinki. All the collected data were confirmed by obtaining the data from all subjects and/or their legal guardians.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: This study was supported by the Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation (2023GXNSFAA026137). This study was supported by grants from Guangxi Health and Family Planning Commission self-funded research project (ZA20230687). This study was supported by Guangxi Medical and health key discipline construction project.
References
- Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol. 2019;317:H891-922.
- [CrossRef] [PubMed] [Google Scholar]
- Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol. 2019;73:89-99.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am J Physiol Heart Circ Physiol. 2018;315:H1341-52.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial dysfunction and myocardial ischemia-reperfusion: Implications for novel therapies. Annu Rev Pharmacol Toxicol. 2017;57:535-65.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481-90.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial quality control in cardiac ischemia/reperfusion injury: New insights into mechanisms and implications. Cell Biol Toxicol. 2023;39:33-51.
- [CrossRef] [PubMed] [Google Scholar]
- Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management. Pharmacol Ther. 2018;191:1-22.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020;36:101607.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNA-3148 inhibits glioma by decreasing DCUN1D1 and inhibiting the NF-kB pathway. Exp Ther Med. 2022;23:28.
- [CrossRef] [PubMed] [Google Scholar]
- OTOP2, Inversely modulated by miR-3148, inhibits CRC cell migration, proliferation and epithelialmesenchymal transition: Evidence from bioinformatics data mining and experimental verification. Cancer Manag Res. 2022;14:1371-84.
- [CrossRef] [PubMed] [Google Scholar]
- Microarray analysis and detection of MicroRNAs associated with chronic thromboembolic pulmonary hypertension. Biomed Res Int. 2017;2017:8529796.
- [CrossRef] [PubMed] [Google Scholar]
- Roles of Krüppel-Like Transcription factors KLF6 and KLF15 in proximal tubular metabolism. Nephron. 2023;147:766-8.
- [CrossRef] [PubMed] [Google Scholar]
- Krüppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest. 2015;125:1347-61.
- [CrossRef] [PubMed] [Google Scholar]
- Krüppel-like factor 6 is a transcriptional activator of autophagy in acute liver injury. Sci Rep. 2017;7:8119.
- [CrossRef] [PubMed] [Google Scholar]
- Declaration of Helsinki: Ethical principles for medical research involving human subjects France: World Medical Association; 2024.
- [Google Scholar]
- Incidence and case-fatality of stroke on the East border of the European union: The Grodno Stroke Study. Stroke. 2010;41:2726-30.
- [CrossRef] [PubMed] [Google Scholar]
- Interactions of trace amines with dopamine in rat striatum. Prog Neuropsychopharmacol. 1981;5:577-80.
- [CrossRef] [PubMed] [Google Scholar]
- Congestive heart failure, hypertension, and hyperreninemia in bilateral Wilms' tumor: Successful medical management. Med Pediatr Oncol. 1986;14:63-6.
- [CrossRef] [PubMed] [Google Scholar]
- miR-3148 Is a novel Onco-microRNA that potentiates tumor growth in vivo. Anticancer Res. 2018;38:5693-701.
- [CrossRef] [PubMed] [Google Scholar]
- The prognostic value of circulating microRNAs in heart failure: Preliminary results from a genome-wide expression study. J Cardiovasc Med (Hagerstown). 2015;16:431-7.
- [CrossRef] [PubMed] [Google Scholar]
- A novel strategy for real-time and in situ detection of cytochrome c and caspase-9 in Hela cells during apoptosis. Analyst. 2014;139:2499-506.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular and immunohistochemical expression of apoptotic proteins Bax, Bcl-2 and Caspase 3 in infantile hemangioma tissues as an effect of propranolol treatment. Immunol Lett. 2017;185:27-31.
- [CrossRef] [PubMed] [Google Scholar]
- The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants (Basel). 2019;8:454.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J Cell Physiol. 2019;234:8122-33.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial quality control: A pathophysiological mechanism and therapeutic target for stroke. Front Mol Neurosci. 2021;14:786099.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981-91.
- [CrossRef] [PubMed] [Google Scholar]
- The PINK1-Parkin axis: An overview. Neurosci Res. 2020;159:9-15.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136:388-402.
- [CrossRef] [PubMed] [Google Scholar]
- LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016;30:3961-78.
- [CrossRef] [PubMed] [Google Scholar]
- The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571-80.
- [CrossRef] [PubMed] [Google Scholar]
- Kruppel-like factor 6 promotes macrophage inflammatory and hypoxia response. FASEB J. 2020;34:3209-23.
- [CrossRef] [PubMed] [Google Scholar]
- Podocyte-Specific Loss of Krüppel-Like factor 6 increases mitochondrial injury in diabetic kidney disease. Diabetes. 2018;67:2420-33.
- [CrossRef] [PubMed] [Google Scholar]
- Klf6 aggravates myocardial ischemia/reperfusion injury by activating Acsl4-mediated ferroptosis. Kaohsiung J Med Sci. 2023;39:989-1001.
- [CrossRef] [PubMed] [Google Scholar]
- WTAP-mediated m6a modification of klf6 aggravates hypoxia/reoxygenation-induced human cardiomyocyte injury. Shock. 2024;62:201-7.
- [CrossRef] [PubMed] [Google Scholar]




