


Translate this page into:
Geniposide alleviates post-myocardial infarction-induced pyroptosis by modulating the thioredoxin-interacting protein/NLRP3 signaling pathway

Honghong He

Ya Liu
*Corresponding authors: Honghong He, Department of Cardiovascular Medicine, The People’s Hospital of Yubei District of Chongqing, Chongqing, China. hehonghong111@163.com
Ya Liu, Department of Cardiovascular Medicine, The People’s Hospital of Yubei District of Chongqing, Chongqing, China. 13983043106@139.com
-
Received: ,
Accepted: ,
How to cite this article: Jiang Y, Su Y, Li C, Jiang W, Wei Y, Chang G, et al. Geniposide alleviates post-myocardial infarction-induced pyroptosis by modulating the thioredoxin-interacting protein/NLRP3 signaling pathway. CytoJournal. 2024;21:80. doi: 10.25259/Cytojournal_139_2024
Abstract
Objective:
Geniposide (GP) provides myocardial cells with protection against pyroptosis-induced damage. However, the mechanisms governing GP’s effect on the thioredoxin-interacting protein (TXNIP)/nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) signaling pathway remain unclear. This study aimed to explore how GP alleviates post-myocardial infarction (MI)-induced pyroptosis through regulation of the TXNIP/NLRP3 pathway.
Material and Methods:
In vivo studies: MI models were established, mouse body weight, heart rate, and blood glucose levels were monitored, and methods, such as cardiac ultrasound, hematoxylin–eosin staining, triphenyltetrazolium chloride staining, terminal deoxynucleotidyl transferase 2'-deoxyuridine 5'-triphosphate nick end labeling staining, quantitative polymerase chain reaction (qPCR), and Western blot (WB), were used to explore the effect of GP on myocardial cell pyroptosis. We explored the role of NLRP3 in GP’s antimyocardial cell pyroptosis through qPCR, WB, immunofluorescence, enzyme-linked immunosorbent assay (ELISA), and other methods. In vitro studies: A chronic hypoxia (CH) cell model was established, and detection methods, such as cell counting kit-8 assay, transmission electron microscopy, ELISA, and immunological assays, were used to explore the effects of GP on CH myocardial cell pyroptosis and GP’s inhibition of the TXNIP/NLRP3 signaling pathway to resist CH myocardial cell pyroptosis.
Results:
In vivo studies revealed that after the treatment with GP, the infarct area of mice’s hearts significantly decreased, cardiac structure and function notably improved, fibroblast proliferation in cardiac tissues decreased significantly, and the pyroptosis level of myocardial cells decreased. GP treatment significantly downregulated the expression levels of type I collagen (Col I), Col III, TXNIP NLRP3, caspase-1, and gasdermin D N-terminal (GSDMD-N). The inhibition of NLRP3 also reduced the expressions of NLRP3, TXNIP, caspase-1, and GSDMD-N in the cardiac tissue, which is concomitant with a decline in reactive oxygen species (ROS) production. In addition, in vitro studies unveiled that GP effectively alleviated pyroptosis in CH myocardial cells, reducing pyroptosis rates, interleukin (IL)-1β, IL-18, lactate dehydrogenase, and creatine kinase-muscle/brain levels. This protective effect was achieved by inhibiting the TXNIP/NLRP3 signaling pathway.
Conclusion:
GP greatly diminishes the extent of infarcted myocardial tissue and mitigates pyroptosis, which improves cardiac structure and function through modulation of the TXNIP/NLRP3 pathway. Furthermore, the inhibition of NLRP3 lowers the expressions of factors associated with pyroptosis in the cardiac tissue and reduces ROS production.
Keywords
Geniposide
NLRP3 protein
Thioredoxin-interacting protein
Myocardial infarction
Pyroptosis

INTRODUCTION
Myocardial infarction (MI) represents a consequential outcome of myocardial ischemia, which results in cell death. This outcome is highly important and should not be overlooked.[1] Myocardial ischemia leads to insufficient oxygen supply to cardiac muscles, which triggers myocardial hypoxia, culminates in apoptotic and necrotic cell death, and ultimately manifesting as MI.[2] MI induces a robust inflammatory response, which fosters cell death, cardiac dysfunction, and alterations in the ventricular remodeling.[3] Further, exploration of this phenomenon revealed that pyroptosis, also known as cell inflammatory necrosis, plays an instrumental role in triggering the inflammatory cascade following MI.[4,5] Pyroptosis is characterized by continuous cell enlargement, which culminates in cell membrane rupture, triggers the release of intracellular contents, and induces an inflammatory reaction.[6] This process is primarily triggered by the inflammasome-mediated activation of caspase-1 and other caspases and subsequent cleavage of gasdermin D (GSDMD).[7] GSDMD, a pore-forming protein within the GSDMD family, functions as an essential initiator of pyroptosis.[8] Thioredoxin-interacting protein (TXNIP) interacts with nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) and participates in inflammasome activation and subsequent pyroptosis.[9] When cells are stimulated by danger signals, excessive reactive oxygen species (ROS) production generates TXNIP, which is originally bound to thioredoxin (TRX), to dissociate and bind to the leucine-rich repeat domain of NLRP3, which initiates and activates the NLRP3 inflammasome.[10-13] This process promotes the generation of active caspase-1, GSDMD, interleukin (IL)-18, and IL-1β.[14] The release of these cytokines exacerbates cellular damage and inflammation. Pyroptosis following myocardial ischemia shows a close association with the severity of myocardial injury. On one hand, cytokines and inflammatory mediators released during pyroptosis further damage on the surrounding myocardial tissue, which exacerbates ischemic injury. On the other hand, pyroptosis adversely affects myocardial regeneration and repair processes, which hinders the recovery of damaged myocardial tissue. Therefore, the regulation of pyroptosis holds important clinical implications.
Recent studies have indicated that certain natural products can potentially inhibit cell pyroptosis and reduce myocardial ischemic damage. Geniposide (GP) protects myocardial cells from pyroptotic damage.[15] This compound is a primary active component of Gardenia jasminoides Ellis[16,17] and possesses diverse pharmacological properties, including antioxidant stress,[18] anti-inflammatory properties,[19] anti-apoptotic effects,[20] antithrombotic activity,[21] anti-endoplasmic reticulum stress,[22] and antifungal properties.[23] Our research group previously reported that GP, through the activation of glucagon-like peptide-1 receptors, inhibits oxidative stress in hypoxia-reoxygenation myocardial cells (reducing ROS/reactive nitrogen species), enhances mitochondrial membrane potential, reduces mitochondrial calcium overload, and improves mitochondrial dysfunction.[18] Moreover, to further explore the role of GP in ischemia-reperfusion injury (RI), our group established a MI/RI model in our preliminary work. Pretreatment with GP considerably improved cardiac function and structure in MI/RI rats and reduced the myocardial cell pyroptosis rate.[24] However, our group did not investigate the effects of GP on inflammation and cell pyroptosis following MI nor the specific mechanisms of GP in the TXNIP/NLRP3 signaling pathway. Therefore, this study aimed to examine the influence of GP on the cell pyroptosis triggered by MI and elucidate its mechanisms of action. We used animal and cell models to evaluate the inhibitory effects of GP on myocardial cell pyroptosis in vivo and in vitro and assessed changes in the TXNIP/NLRP3 signaling pathway. This work also elucidated the regulatory role of GP in the TXNIP/NLRP3 signaling pathway in myocardial ischemic damage and provides new therapeutic strategies and drug targets for post-MI treatment.
MATERIAL AND METHODS
Experimental animals
Our study included 64 male 8-week-old specific pathogen free (SPF)-grade C57BL/6J wild-type mice, with mouse each weighing between 18 and 20 g (Chongqing Ensiweier Biological Technology Co., Ltd). In addition, our animal facilities adhered to a 12 h light-dark cycle, which ensured the unrestricted access of mice to water and food. Ambient temperature was kept between 23°C and 25°C. After a 1-week acclimation period, participation of the mice in the experiment commenced.
All animal procedures employed in the project were approved by Institutional Animal Care and Use of Chongqing Medical University, Approval number: IACUC-CQMU-2023-0315.
Experimental model
The anesthetic procedure for our MI model group mice was conducted as follows: We intraperitoneally injected the mice with 50 mg/kg sodium pentobarbital (0.3%) to ensure that they were anesthetized. Subsequently, a custom-made intubation device was inserted into the trachea to assist in ventilation. The mice were securely fastened to the surgical platform with their chests depilated. Next, thoracotomy was performed on the left side between the third and fourth ribs to expose the heart. A suture needle was then passed through the ligature to ligate the root of the left anterior descending coronary artery. Finally, the thoracic cavity was promptly closed, and suturing was performed.
The control group (sham) underwent the following procedures: A similar MI model operation was performed, but thoracotomy was conducted without ligating the root of the left anterior descending coronary artery.
Group dosing and sampling of experimental animals
Experimental Assessment of GP’s Effect on Post-MI cardiac pyroptosis:
Control group: Following surgery, daily oral administration of 0.2 mL saline was continued for 14 consecutive days.
MI Group: Postsurgery, daily oral administration of 0.2 mL saline was sustained for 14 consecutive days.
MI+low-dose GP Group (MI + L + GP): After surgery, daily oral administration of GP at 50 mg/kg (drug concentration 5 mg/mL, 0.2 mL/dose) was maintained for 14 days.
MI+high-dose GP Group (MI + H + GP): Postsurgery, daily oral administration of GP at 100 mg/kg (drug concentration 10 mg/mL, 0.2 mL/dose) was continued for 14 consecutive days (8 mice per group).
-
Experimental evaluation of NLRP3’s Role in GP-mediated pyroptosis inhibition
Control Group: An open-chest surgery was performed with the anterior descending branch ligation omitted. Postoperatively, saline was orally administered on a daily basis.
MI: Following MI induction, saline was orally administered daily.
MI+GP group: After the induction of MI, daily oral administration of GP at 100 mg/kg was administered.
MI+GP+NLRP3 inhibitor (MCC950) group (MI + GP + MCC950): After MI induction, daily oral administration of GP (100 mg/kg) was combined with intraperitoneal injection of 10 mg/kg MCC950 (A421651-0002, Sangon Biotech, Shanghai, China) (8 mice per group).
After a 14-day period, euthanasia was performed using high-concentration CO2. Subsequently, blood serum was collected, and further analysis involved the retrieval of the heart and lungs tissues.
Mouse physiological and cardiac structure and function assessment
We conducted measurements on various groups of mice at four different time points on days 0, 3, 7, and 14. These measurements included monitoring of the body weight, heart rate, and blood glucose levels. In addition, the relative weights of the heart, wet lung, and dry lung in each group of mice were calculated. Furthermore, cardiac ultrasonography was performed to assess the structural and functional aspects of the heart. Transthoracic echocardiography was performed using a VividTM E95 echocardiogram system (GE Healthcare, Boston, MA, USA). The mice were anesthetized with 2% isoflurane and positioned on the echo platform in the left lateral decubitus position. To maintain their core body temperature, we heated the platform to 37°C. After placement, the fur on the chest was gently removed using a hair removal cream. Heart rate, which typically ranges 400 bpm–500 bpm, was determined from cardiac cycles. L8-18i-D PROBE ultrasound probe (GE Healthcare, Boston, MA, USA) was placed in parasternal view at the level of papillary muscles to capture short-axis images of the heart. M-Mode imaging was then performed to measure the end systolic volume (ESV), end diastolic volume (EDV), left ventricular internal diameter at systole (LVIDs), and left ventricular internal diameter at diastole (LVIDd). The left ventricular ejection fraction (EF) was calculated as (EDV − ESV)/EDV, and the left ventricular fractional shortening (FS) was (LVIDd − LVIDs)/LVIDd. All measurements were conducted by an investigator who was unaware of the treatment conditions.
Triphenyltetrazolium chloride (TTC) staining
The TTC staining method was employed to assess the extent of MI in the left ventricular tissue of mouse hearts. Freshly harvested mouse myocardial tissues were rinsed with sterile saline (ST341–500 mL, Beyotime, Shanghai, China) and then frozen at −20°C for 20 min. After freezing and starting from the apex, the tissues were serially sliced into thin sections with thicknesses not exceeding 1 mm. These sections were then immersed in 2% TTC staining solution (Sigma, St. Louis, Missouri, USA) and incubated at 37°C in a light-protected environment for 20 min. Following incubation, the tissues were fixed with 4% paraformaldehyde (p0099, Beyotime, Shanghai, China), and photographs were captured after 8 h. The infarcted myocardial area was calculated using Image-Pro Plus Version 6.0 software (Media Cybernetics, Rockville, Maryland, USA).
Hematoxylin–eosin (HE) staining
HE staining was performed to determine the overall structure and inflammation status of the left ventricular tissue in mouse hearts. Initially, left ventricular tissue samples were prepared by cleaning and fixing the tissue sections. Subsequently, a series of procedures, including dehydration, transparency, and paraffin embedding, was performed to create paraffin sections. The paraffin sections were rehydrated to restore their aqueous state. For specific staining effects, we conducted HE staining using G1004 (Servicebio, Wuhan, China) and G1002 (Servicebio, Wuhan, China) to highlight distinct cell components. Then, ethanol dehydration and transparency treatments were conducted, followed by reinforcement of the sections with neutral resin. Then, images of the stained sections were captured using an optical microscope (MF53, Mshot-Mingmei, Guangzhou, China). This sequence of steps facilitated the observation and documentation of the microstructure and inflammatory conditions of the cardiac tissue.
Terminal deoxynucleotidyl transferase (TdT) 2’-deoxyuridine 5’-triphosphate nick end labeling (TUNEL) staining
TUNEL assay was performed to investigate pyroptosis in myocardial cells. During the experiment, we used a Diaminobenzidine (DAB) (Streptavidin-horse radish peroxidase [SA-HRP]) TUNEL Cell Apoptosis Detection Kit (G1507, Servicebio, Wuhan, China) to assess cardiac cell pyroptosis. After deparaffinization and rehydration of the paraffin sections, incubate the sections with proteinase K working solution for 20 min, Equilibration Buffer for 10 min, TdT incubation buffer for 1 h, and StreptavidinHRP reagent for 30 min. After each incubation step, wash the samples 3-4 times with phosphate buffered saline (PBS) solution, 5 min each time. Then, we performed DAB staining (ZLI-9019; ZSGB-BIO, Beijing, China) and observed the staining patterns. Next, the sections were stained with hematoxylin solution (G1004, Servicebio, Wuhan, China) for 1 min, thoroughly washed with distilled water, differentiated briefly in hematoxylin differentiation solution for approximately 2 s, immediately rinsed with distilled water, washed with tap water for 10 min to achieve bluing, and finally rinsed with distilled water. After staining, the sections were dehydrated and mounted. Finally, the sections and capture images were observed using an inverted microscope (Mshot MF53, Mshot-Mingmei, Guangzhou, China). The apoptosis rate was calculated using ImageJ (version 1.53, Wayne Rasband, National Institutes of Health, https://imagej.nih.gov/ij/), with the average apoptosis rate being the mean of apoptosis rates from three images.
Quantitative polymerase chain reaction (qPCR)
During this experimental phase, we employed qPCR to assess the expressions of type I collagen (Col I), Col III, TXNIP, and NLRP3 mRNA, with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) serving as the internal reference. Relative expressions, which indicate fold differences in the target gene expressions between the experimental and control groups, were assessed through the 2−△△Ct method. Table 1 lists the polymerase chain reaction primer sequences.
| Primers | Sequences |
|---|---|
| Col I-F | GCAAGAGGCGAGAGAGGTTT |
| Col I-R | GACCACGGGCACCATCTTTA |
| Col II-F | GAGGAATGGGTGGCTATCCG |
| Col II-R | TCGTCCAGGTCTTCCTGACT |
| TXNIP-F | CAATCAGTAGGCGGGTCTCC |
| TXNIP-R | TGTTCTGACATCCACCCAGC |
| NLRP3-F | AAGGCTGCTATCTGGAGGAACT |
| NLRP3-R | ATACAGCCTTTCTCGGGCG |
| m-GAPDH-F | GGAGAGTGTTTCCTCGTCCC |
| m-GAPDH-R | TTTGCCGTGAGTGGAGTCAT |
Col I: Type I collagen, Col III: Type III collagen, TXNIP: Thioredoxin interacting Protein, NLRP3: NOD-like receptor protein 3, GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
Western blot (WB)
WB assay was conducted to evaluate the protein levels of TXNIP, NLRP3, caspase-1, and GSDMD N-terminal (GSDMD-N) in myocardial tissue. Initially, a small quantity of cardiac tissue sample was placed in 2 mL grinding tubes, and radioimmunoprecipitation assay lysis buffer (P0013B, Beyotime, Shanghai, China) was added to extract proteins. Protein concentration was quantified through the bicinchoninic acid assay. The protein samples were then combined with ×5 sodium dodecyl sulfate (SDS) loading buffer (G2083, Servicebio, Wuhan, China), heated in a boiling water bath to denature, and separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE). The gel was transferred onto a polyvinylidene fluoride membrane (10600023, Cytiva, Shanghai, China), which was then placed in Tris buffered saline with Tween 20 (TBST) buffer and blocked with 5% skim milk at room temperature for 1 h. Next, primary antibodies, including TXNIP (1:1,000), NLRP3 (1:1,000), caspase-1 (1:1,000), GSDMD-N (1:1,000), and GAPDH (1:1,000), were used (ABclonal Company in Wuhan, China; A9342, A5652, A0964, A20197, and A19056, respectively). Incubation with the primary antibodies was performed overnight on a shaker at 4°C. Subsequently, the secondary antibody, horse radish peroxidase (HRP) goat antirabbit Immunoglobulin G IgG (H + L) (AS014, ABclonal, Wuhan, China), was diluted at a ratio of 1:2,000 in TBST and incubated at room temperature for 1 h. Immunoreactivity was detected using an enhanced chemiluminescence reagent (34580, Thermo, Massachusetts, USA) and imaged using a nucleic acid and protein gel imaging system (Universal Hood II, Bio-Rad, CA, USA). The grayscale values of the bands were analyzed using ImageJ (version 1.53, Wayne Rasband, National Institutes of Health, https://imagej.nih.gov/ij/). This analysis determined the relative protein expressions through calculation of the ratio of the target protein’s grayscale value to that of GAPDH.
Immunohistochemistry (IHC)
IHC was used to detect the protein levels of Col I and Col III in myocardial tissue. The involved procedure included deparaffinization and rehydration of paraffin sections, antigen retrieval by boiling, treatment with 3% hydrogen peroxide, PBS washing, incubation with the blocking serum, overnight primary antibody (1:200), secondary antibody (1:50), DAB chromogen staining, counterstaining with hematoxylin, differentiation, dehydration with ethanol and xylene, and mounting with neutral resin. The slides were examined for blue-stained nuclei and brown-yellow positive areas under a microscope. Imaging was conducted using an Mshot MF53 inverted microscope (Guangzhou Mingmei Optoelectronics Technology Co., Ltd).
Primary antibody: Col I/COL1A1 rabbit pAb (A1352, ABclonal, Wuhan, China), Col III antibody (Orb10437, Biorbyt, Wuhan, China); secondary antibody: HRP-labeled goat anti-rabbit IgG (H + L) (A0208, Beyotime, Shanghai, China).
Immunofluorescence (IF)
IF method was used to assess the expression and localization of TXNIP and NLRP3 proteins in the myocardial tissue. Paraffin-embedded sections were initially deparaffinized and rehydrated, subjected to antigen retrieval through boiling for 30 min, and allowed to cool to room temperature. Subsequently, the sections were incubated for 30 min with a goat serum at room temperature for blocking, followed by the removal of excess blocking solution without rinsing. The sections were subsequently incubated overnight at 4°C with primary antibodies targeting NLRP3 (R381207, Zenbio, Chengdu, China) and TXNIP (R382206, Zenbio, Chengdu, China) at a 1:200 dilution. Following primary antibody incubation, a secondary antibody solution, Cy3 goat antirabbit IgG (H + L) (AS007, ABclonal, Wuhan, China) diluted at 1:200, was applied and treated at room temperature for 1.5 h under light-shielded conditions. Next, 4’,6-diamidino-2-phenylindole (DAPI) staining was carried out through the addition of DAPI solution and incubated for 5 min under light protection to stain the cell nuclei. The cells were rinsed with PBS to remove excess DAPI. Finally, all sections were covered with an autofluorescence quenching agent, and images were captured using an inverted microscope (Mshot MF53, Mshot-Mingmei, Guangzhou, China).
Enzyme-linked immunosorbent assay (ELISA)
The ROS levels in mouse myocardial tissue were assessed using a mouse ROS ELISA Kit (CB10366-Mu, COIBO BIO, Shanghai, China).
Cell culture
Isolation of primary cardiomyocytes:[25] Hearts were harvested from 1 to 3-day-old SPF-grade C57BL/6J wild-type mice. The chest was disinfected with 70% ethanol and opened along the left sternum, and the heart was removed. The heart was rinsed with cold PBS to remove the atria and blood vessels. The rinsed heart tissue was then placed in 0.06% trypsin (S310KJ, Basal media, Shanghai, China) and incubated at 4°C for 5 h to loosen the tissue. The heart tissue was then transferred to a centrifuge tube with a digestion solution of 0.1% Col II (17101015, Thermo, Massachusetts, US) and 0.5% bovine serum albumin, cut into 1 mm3 fragments, and incubated for digestion. The supernatant was collected, and the cells were seeded into culture dishes. After differential adherence, cells from the culture medium were collected, counted, adjusted to a density of 1 × 105 cells/mL, and then seeded into a six-well plate. Cell morphology and contraction were observed under an inverted microscope, and before the experiments, the purity of cardiomyocytes was confirmed by α-actin fluorescence ≥90%. Cell lines underwent regular mycoplasma contamination screening and were evaluated through short-tandem repeat DNA profiling.
Establishment of the chronic hypoxia (CH) model
Primary cardiomyocytes were seeded into cell culture flasks or plates and cultured until 70–80% confluence. The cells were washed thrice with serum-free Dulbecco’s Modified Eagle Medium (DMEM)/F12 (C11995500BT, GIBCO, Massachusetts, US) and then incubated with 4 mL serum-free DMEM/F12 in a hypoxic chamber (1% oxygen, 5% Carbon dioxide) at 37°C for an appropriate period to induce hypoxia. After 48 h of hypoxia, the CH model was established.
Virus construction and cell transfection
The lentivirus p-Receiver-TXNIP was produced using full-length human TXNIP complementary deoxyribonucleic acid segment which was cloned into the lentiviral vector p-Receiver-Lv185, and 293T cells (CRL11268, ATCC, Virginia, US). When cardiomyocytes reached 70–80% confluence in culture, they were infected with the lentivirus.
Cell experiment groups
Investigation of the effect of GP on CH-induced cardiomyocyte apoptosis:
Control group: Primary cardiomyocytes cultured under normal conditions.
Control + GP group: GP added at a concentration of 40 µM to the cardiomyocyte culture under normal conditions.
CH group: Cardiomyocytes cultured to 70–80% confluence and subjected to the CH model established through the method described above.
GP + CH group: GP added at a concentration of 40 µM to the cardiomyocyte culture for 30 min of pretreatment followed by establishment of the CH model.
Investigation of whether GP attenuates CH-induced cardiomyocyte apoptosis by inhibiting the TXNIP/NLRP3 signaling pathway:
Control group: Cultured in blank culture medium.
CH + overexpression-negative control (oe-NC) group: Cardiomyocytes cultured to 70–80% confluence and subjected to the CH model.
CH + -TXNIP group: Cardiomyocytes infected with lentivirus overexpressing TXNIP followed by establishment of the CH model.
CH + OE-TXNIP + GP group: Cardiomyocytes infected with lentivirus overexpressing TXNIP and treated with GP followed by establishment of the CH model.
Cell counting kit-8 (CCK-8) assay
Using the same batch of cardiomyocytes, the cell suspension density was adjusted to 1 × 105 cells/mL. Then, 100 µL of this suspension was dispensed into each well of a 96-well plate, with five replicate wells per group. The plate was placed in a standard cell culture incubator, and the cells were incubated until they reach 70–80% confluence. The cells were treated in accordance with each group’s requirements. Then, the culture medium was replaced with 100 µL CCK-8 (C0038, Beyotime, Shanghai, China) solution per well and incubated in a 37°C cell culture incubator for 2 h. The 96-well plate was transferred to a microplate reader (CMax Plus, Molecular Devices, California, US), the instrument parameters were set, and the absorbance was measured at 450 nm for each well. The cell viability for each group was determined using the recorded absorbance values.
Transmission electron microscopy (TEM)
Observation of cell necrosis morphology through electron microscopy revealed the following: enlarged, deformed, and swollen cells, ruptured membrane, distorted organelles, irregular DNA degradation, and intact nuclei. Cells were collected, washed with PBS, digested with trypsin, and centrifuged, and fixed in centrifuge tubes containing glutaraldehyde and osmium tetroxide. Sequential dehydration with acetone was performed, followed by infiltration and embedding in epoxy resin (Epon 812). Ultrathin sections approximately 50 nm thick were prepared, followed by double staining (uranyl acetate and lead citrate), and observed through TEM (JEM-1400PLUS, JEOL, Tokyo Metropolis, Japan).
ELISA
ELISA methods were used to quantify the levels of IL-1β, IL-18, lactate dehydrogenase (LDH), and creatine kinasemuscle/brain (CK-MB) levels were measured using the mouse IL-1β ELISA Kit (EK201B/3–96, MultiSciences [LiankeBio], Hangzhou, China), IL-18 levels with a mouse IL-18 ELISA Kit (EK218–96, MultiSciences [LiankeBio], Hangzhou, China), LDH levels with the mouse LDH ELISA Kit (CB10587-Mu, COIBO BIO, Shanghai, China), and CKMB levels with the mouse CK-MB ELISA Kit (CB10373-Mu, COIBO BIO, Shanghai, China).
Immunoprecipitation (IP)
IP was performed to verify the interaction between TXNIP and TRX, and their association with the NLRP3 inflammasome. Cell proteins were extracted and adjusted to a concentration of 200 µg/mL. IP was performed separately using antibodies against TXNIP (R382206, Zenbio, Chengdu, China) and TRX (ab273877, Abcam, Cambridge, UK). The samples were incubated with the respective antibodies in IP buffer for 12 h, mixed with 20 µL protein A/G agarose beads, and incubated at room temperature for an additional 4 h. After washing the beads with IP buffer, the immunoprecipitated proteins were resuspended in the SDS sample buffer, boiled for 3 min, and analyzed by SDS-PAGE.
Data statistics and analysis
The experimental data acquired in this study are portrayed as mean ± standard error. Group comparisons were executed through the one-way analysis of variance, and data analysis and graphical representation were facilitated through GraphPad Prism 8 software (GraphPad Software Inc., La Jolla, California, USA) (www.graphpad.com). Variances were considered statistically significant whenever a parameter presented P < 0.05.
RESULTS
Effects of GP on cardiac indices, cardiac structure and function, and MI area in MI mice
We monitored the mice at multiple time points, including days 0, 3, 7, and 14, focusing on their weight, heart rate, and blood glucose levels. Our results reveal the significant weight change in the MI group compared with the control group. (P < 0.01), and the heart rate significantly increased on days 0, 7, and 14 (P < 0.05, P < 0.01, P < 0.001, respectively). The blood glucose levels in the MI group also increased on day 14 (P < 0.05). Subsequently, observations were made following GP treatment, which indicate that no significant changes occurred in weight or blood glucose levels across all groups. However, in the MI + L + GP and MI + H + GP groups, the heart rate significantly increased on day 3 (P < 0.001), and it notably decreased on day 14 in MI + H + GP group (P < 0.01) [Figure 1a-c]. No significant differences were observed in wet lung and dry lung relative weights among the groups [Figure 1d and e]. The MI group exhibited a significant increase in relative heart weight (P < 0.01) [Figure 1f]. In addition, the area of heart infarction significantly increased in the MI group (P < 0.001), whereas it markedly decreased in the MI + L + GP, MI + H + GP groups (P < 0.01) [Figure 1g and h]. Ultrasound assessment of heart structure and function revealed a significant decline in EF (Teich) and %FS in MI group (P < 0.001). However, after GP treatment, compared with the MI group, the MI + L + GP and MI + H + GP groups exhibited a significant improvement in EF (Teich) and %FS (P < 0.001). No significant differences were observed in the heart rate among the groups [Figure 1i]. This finding suggests that GP can improve the symptoms of MI in mice.
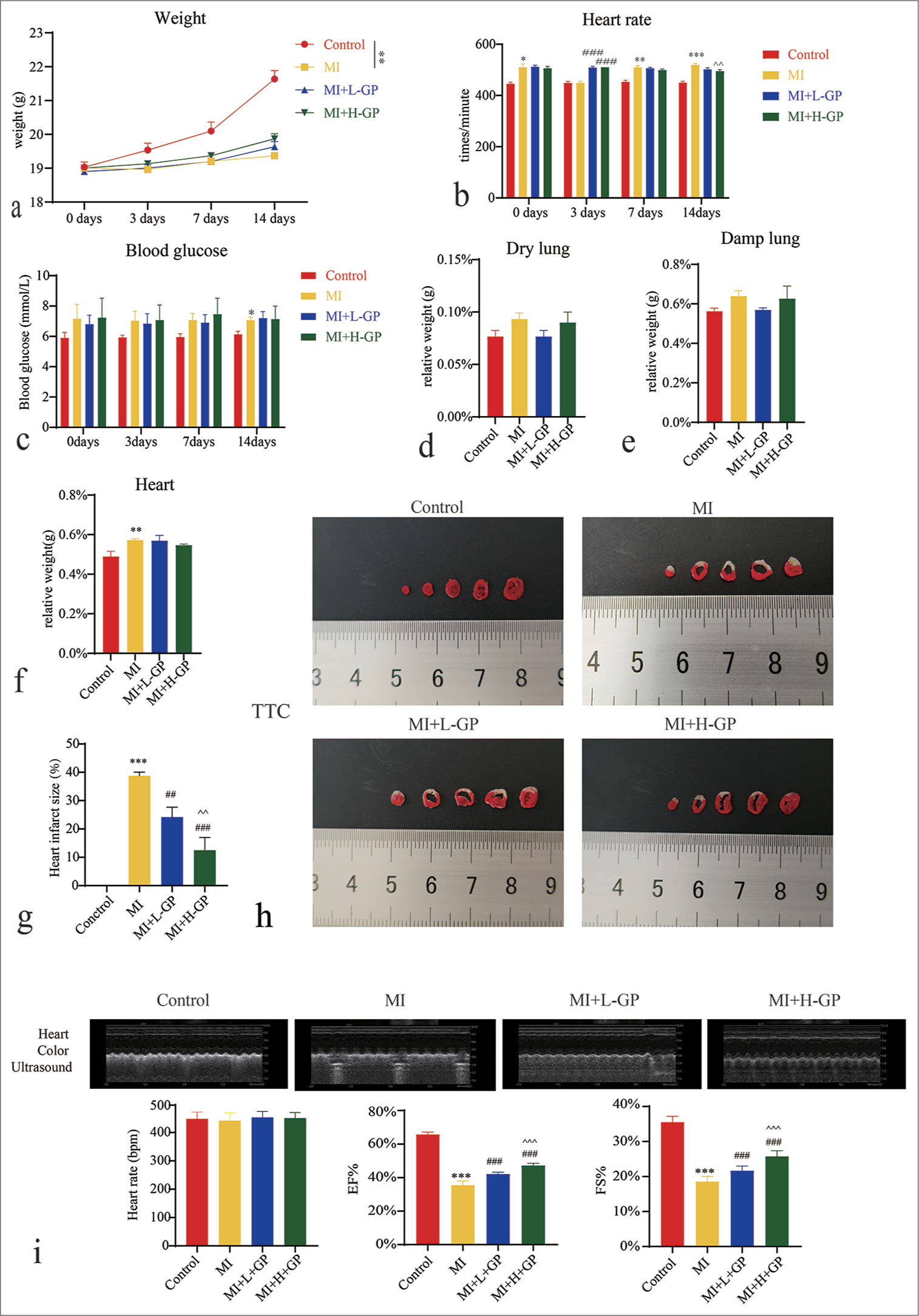
- Effects of Geniposide (GP) treatment on physiological indicators and heart infarct size in myocardial infarction (MI) mice (n = 8). (a) Weight, (b) heart rate, (c) blood glucose, (d) wet lung relative weight, (e) dry lung relative weight, (f) heart relative weight, (g and h) heart infarct size, and (i) heart structure and function. Compared with the control group, ✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001; compared with the MI group, ##P < 0.01, ###P < 0.001; compared with the MI + Low (L)-GP group, ^^P < 0.01, ^^^P < 0.001.
GP’s effects on cardiac histomorphology and cardiomyocyte pyroptosis
Pathological examination of the myocardium revealed the following: In the control group, the myocardial tissue structure showed no evident abnormalities. The myocardial fibers were cord-like and displayed a tight arrangement, with intact myocardial cell structures and distinct boundaries. In the MI group, the orderly arrangement of myocardial fibers was lost and became loosely arranged with a significant increase in fibrous tissue. Damaged, deformed, swollen, and ruptured myocardial cell structures were observed, with noticeable inflammatory cell infiltration. The MI + L + GP group exhibited disorganized myocardial tissue structure, pronounced myocardial cell swelling, reduced inflammatory cell infiltration, and tissue hemorrhage. Compared with the MI group, the extent of damage was considerably alleviated. In the MI + H + GP group, the myocardial fibers were continuously arranged but were slightly loose in structure. Although myocardial cells exhibited swelling, their morphological structure remained largely intact [Figure 2a].
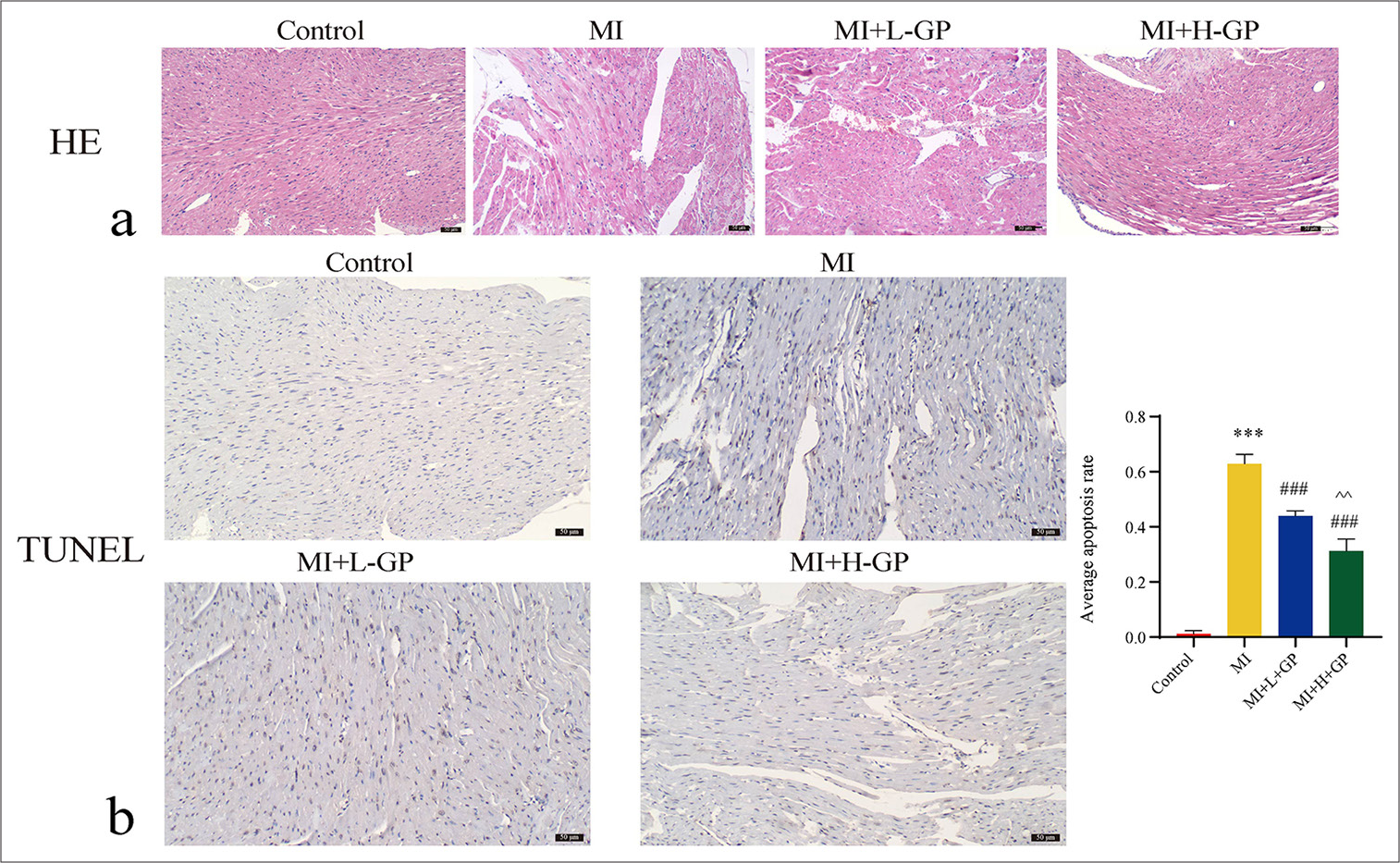
- Myocardial tissues of each group of mice were subjected to hematoxylin–eosin (HE) and terminal deoxynucleotidyl transferase 2’-deoxyuridine 5’-triphosphate nick end labeling (TUNEL) staining (scale bar = 50 µm) (n = 3). (a) HE (×200), (b) TUNEL (×200). Compared with the control group, ✶✶✶P < 0.001; compared with the myocardial infarction (MI) group, ###P < 0.001; compared with the MI + L-Geniposide group, ^^P < 0.01.
TUNEL staining results of the control group displayed minimal positive reactions. However, in the MI group, a substantial increase in the number of TUNEL-positive cells occurred within myocardial cells. In comparison with the MI group, MI + L + GP and MI + H + GP groups exhibited a significant decrease in the TUNEL-positive cell rate (P < 0.001). In comparison with the MI + L-GP group, the MI + H + GP group exhibited a further significant decrease in the TUNEL-positive cell rate (P < 0.01) [Figure 2b]. This result suggests that GP can alleviate MI-induced apoptosis.
Effects of GP on the level of Col I/Col III/pyroptosis-related genes in myocardial tissue
Figure 3a illustrates the effect of GP on the expressions of Col I, Col III, TXNIP, and NLRP3 mRNA in myocardial tissue. The MI group exhibited significant increases in Col I, Col III, TXNIP, and NLRP3 mRNA levels compared with the control group (P < 0.001). By contrast, the MI + L + GP and MI + H + GP groups demonstrated the notably reduced expressions of these markers compared with those of the MI group (P < 0.001). Moreover, the MI+H+GP group showed further significant decreases in the Col I, Col III, TXNIP, and NLRP3 mRNA levels compared with the MI + L + GP group (P < 0.05, P < 0.01, and P < 0.001, respectively).
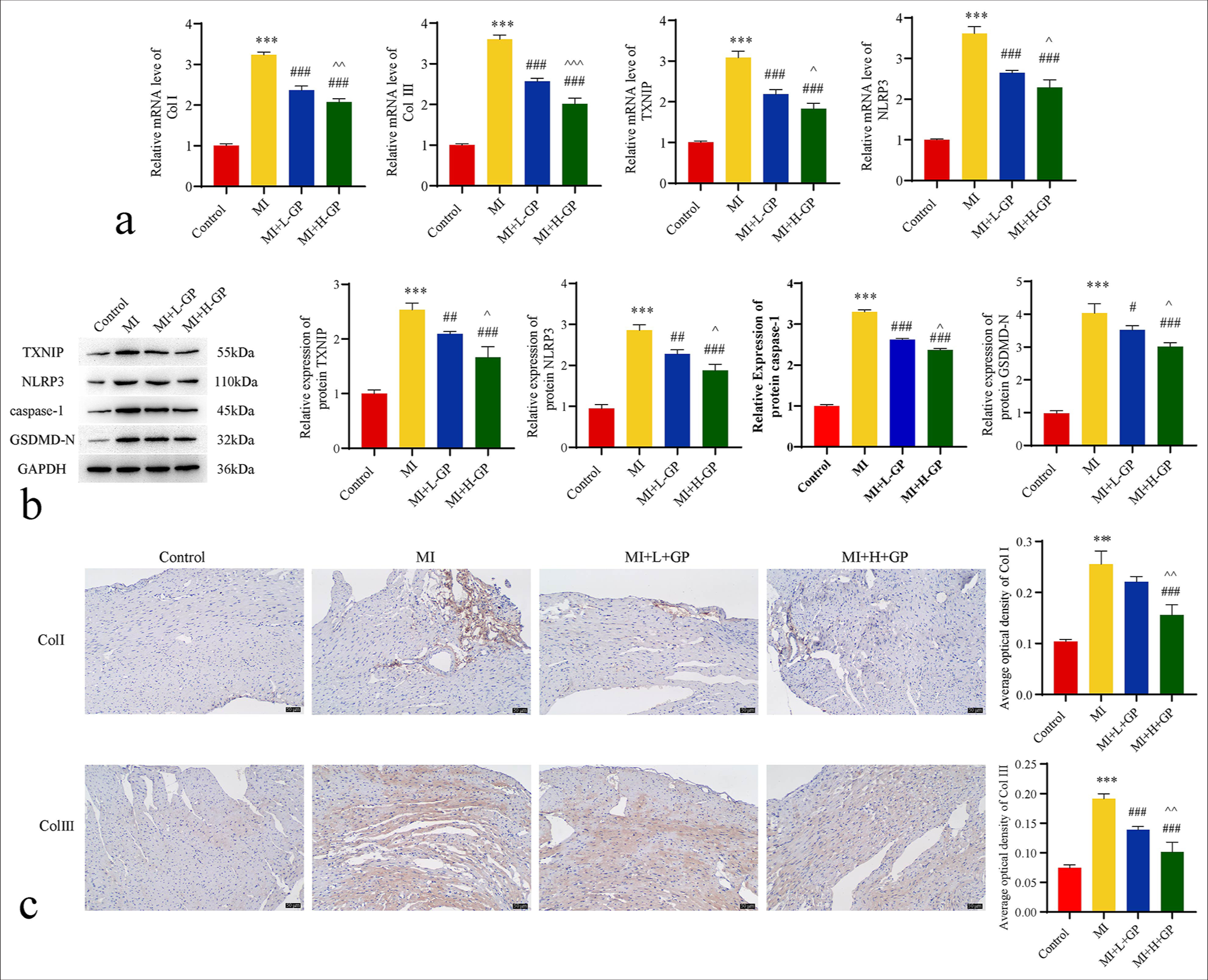
- Expressions of type I collagen (Col I)/type III collagen (Col III)/apoptosis-related genes in mouse myocardial tissues of each group (n = 3). (a) Quantitative polymerase chain reaction assessed the expression of Col I, Col III, thioredoxin-interacting protein (TXNIP), and NLRP3 mRNA. (b) Western blot evaluated protein expressions of TXNIP, NLRP3, caspase-1, and gasdermin D N (GSDMD-N)-terminal. (c) Immunohistochemistry (IHC) staining detected the protein expression levels of Col I and Col III (IHC: ×200, scale bar = 50 µm). Compared with the control group, ✶✶✶P< 0.001; as compared with the myocardial infarction (MI) group, #P < 0.05, ##P < 0.01, ###P < 0.001; as compared with the MI+L-Geniposide (L-GP) group, ^P < 0.05, ^^P < 0.01, ^^^P < 0.001. NLRP3: NOD-like receptor protein 3, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, H-GP: High geniposide.
Figure 3b demonstrates the influence of GP on the levels of TXNIP, NLRP3, caspase-1, and GSDMD-N proteins in myocardial tissue. The MI group showed marked increases in the levels of TXNIP, NLRP3, caspase-1, and GSDMD-N compared with the control group (P < 0.001). By contrast, the MI + L + GP and MI+H+GP groups exhibited significant reductions in these proteins compared with the MI group (P < 0.05, P < 0.01, and P < 0.001, respectively). In addition, the MI+H+GP group displayed a further significant decrease in TXNIP, NLRP3, caspase-1, and GSDMD-N levels compared with the MI + L + GP group (P < 0.05).
Figure 3c illustrates the effect of GP on the levels of Col I and Col III proteins in myocardial tissue. The MI group exhibited a significant increase in Col I and Col III compared with the control group (P < 0.001). However, the MI + H + GP group showed a notable reduction in Col I levels compared with the MI group (P < 0.001). The MI + L + GP and MI + H + GP groups demonstrated significant decreases in Col III levels relative to the MI group (P < 0.001). Furthermore, the MI + H + GP group experienced further significant reductions in Col I and Col III compared with the MI + L + GP group (P < 0.01).
Role of NLRP3 in GP-mediated pyroptosis resistance
We explored the effect of MCC950 on the expression of NLRP3, TXNIP, caspase-1, GSDMD-N, and ROS levels in cardiac tissues [Figure 4].
![Effects of NOD-like receptor protein 3 (NLRP3) inhibitors on the expression of NLRP3, thioredoxin-interacting protein (TXNIP), caspase-1, gasdermin D N-terminal (GSDMD-N), and reactive oxygen species (ROS) levels in cardiac tissue during Geniposide (GP)-mediated anti-pyroptosis (n = 3). (a) mRNA level of NLRP3, TXNIP; (b) expression of caspase-1 and GSDMD-N; (c and d) NLRP3 (Immunofluorescence [IF]: ×400, scale bar = 50 µm) and TXNIP (IF: ×400, scale bar = 50 µm); (e) ROS. Compared with the control group, ✶✶✶P < 0.001; as compared with the myocardial infarction (MI) group, ##P < 0.01, ###P < 0.001; as compared with the MI + GP group, ^P < 0.05, ^^^P < 0.001. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, DAPI: 4’,6-diamidino-2-phenylindole, MCC950: NLRP3 inhibitor.](/content/105/2024/21/1/img/Cytojournal-21-80-g004.png)
- Effects of NOD-like receptor protein 3 (NLRP3) inhibitors on the expression of NLRP3, thioredoxin-interacting protein (TXNIP), caspase-1, gasdermin D N-terminal (GSDMD-N), and reactive oxygen species (ROS) levels in cardiac tissue during Geniposide (GP)-mediated anti-pyroptosis (n = 3). (a) mRNA level of NLRP3, TXNIP; (b) expression of caspase-1 and GSDMD-N; (c and d) NLRP3 (Immunofluorescence [IF]: ×400, scale bar = 50 µm) and TXNIP (IF: ×400, scale bar = 50 µm); (e) ROS. Compared with the control group, ✶✶✶P < 0.001; as compared with the myocardial infarction (MI) group, ##P < 0.01, ###P < 0.001; as compared with the MI + GP group, ^P < 0.05, ^^^P < 0.001. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, DAPI: 4’,6-diamidino-2-phenylindole, MCC950: NLRP3 inhibitor.
In the MI group, myocardial tissue exhibited significantly elevated levels of NLRP3, TXNIP, caspase-1, and GSDMD-N compared with those in the control group (P < 0.001). By contrast, the MI + GP group showed significant reductions in these markers compared with the MI group (P < 0.001, P < 0.01). In addition, the MI + GP + MCC950 group demonstrated a further decrease in NLRP3, caspase-1, and GSDMD-N levels compared with the MI + GP group (P < 0.001, P < 0.05). [Figure 4a-d] provided detailed results. Furthermore, ROS levels in the cardiac tissues of the MI group were significantly higher compared with those of the control group (P < 0.001). The MI + GP group exhibited a substantial reduction in the ROS levels compared with the MI group (P < 0.001). The MI + GP + MCC950 group also demonstrated a significant decrease in the ROS levels (P < 0.001). [Figure 4e] shows the detailed results.
Effect of GP on myocardial cell necrosis induced by CH
In the control + GP group, myocardial cell viability, IL-1β and IL-18 levels and LDH and CK-MB levels remained unchanged compared with the control group (P > 0.05). By contrast, the CH group exhibited a significant reduction in myocardial cell viability (P < 0.01) and notable increases in IL-1β (P < 0.01) and IL-18 (P < 0.001) levels, along with elevated levels of LDH and CK-MB (P < 0.001). Compared with the CH group, the GP + CH group showed a significant improvement in myocardial cell viability (P < 0.05) and a marked decrease in IL-1β and IL-18 (P < 0.05) levels and lower LDH (P < 0.01) and CK-MB (P < 0.001) levels [Figure 5a-c]. The mitochondria of CH group cells revealed significant shrinkage and deformation, membrane rupture, and matrix leakage, with an enlarged intermembrane space of intact mitochondria, which indicates severe necrosis. After GP treatment, the mitochondria displayed deformation, membrane rupture, and matrix leakage but not the enlargement of the intermembrane space. These findings suggest that although GP cannot completely reverse the effects of necrosis, it can alleviate CH-induced myocardial cell necrosis to some extent [Figure 5d].

- Effect of geniposide (GP) on myocardial cell necrosis induced by chronic hypoxia (CH) (n = 3). (a) Cell counting kit-8 detected cell viability; (b and c) enzyme-linked immunosorbent assay-detected interleukin (IL)-1β, IL-18, lactate dehydrogenase, and CK-MB; (d) Transmission electron microscopy-detected cell pyroptosis. N: Nucleus, Mi: Mitochondria, ASS: Autophagolysosome, RER: Rough endoplasmic reticulum, red arrow: Mitochondrial membrane rupture, green arrow: Mitochondrial deformation, yellow arrow: Enlargement of the mitochondrial intermembrane space. Compared with the control group, ✶✶P < 0.05, ✶✶✶P < 0.001; as compared with the CH group, ##P < 0.01, ###P < 0.001. Compared with the control group; “ns” stands for no significant difference. CK-MB: Creatine kinase-myocardial band levels, LDH: Lactate dehydrogenase.
GP inhibits TXNIP/NLRP3 signaling pathway to counter CH-induced myocardial cell necrosis
Compared with the control group, the CH + oe-NC group showed a significantly reduced myocardial cell viability (P < 0.001), accompanied with elevated levels of IL-1β (P < 0.01), IL-18 (P < 0.01), LDH (P < 0.001), and CKMB (P < 0.001). The CH + oe-TXNIP group revealed further reduction in myocardial cell viability (P < 0.001), alongside even higher levels of IL-1β (P < 0.01), IL-18 (P < 0.05), LDH (P < 0.001), and CK-MB (P < 0.001). These results imply that oe-TXNIP exacerbated the myocardial cell necrosis induced by CH. Compared with the CH + oe-TXNIP group, the GP + CH + oe-TXNIP group presented a significantly increased myocardial cell viability (P < 0.001), accompanied with decreased levels of IL-1β (P < 0.001), IL-18 (P < 0.01), LDH (P < 0.001), and CK-MB (P < 0.001) [Figure 6a-c]. Electron microscopy revealed no abnormal morphology and clear internal structure in the control group. In the CH + oe-NC group, the cells exhibited considerable mitochondrial shrinkage and deformation, membrane rupture, matrix leakage, and a large number of autophagic lysosomes. The CH + oe-TXNIP group displayed extensive mitochondrial rupture and signs of shrinkage in the intact mitochondria, with blurred membrane structures and an increase in autophagic lysosomes. However, after GP treatment, only a few mitochondria showed rupture, mitochondrial shrinkage disappeared, and the content of autophagic lysosomes decreased, which indicate the reduced degree of cell damage compared with the first two groups. This result suggests that GP can alleviate CH-induced cell necrosis [Figure 6d]. Finally, we performed IP to identify the interaction between TXNIP and NLRP3 in myocardial cells. GP substantially reduced the interaction between TXNIP and NLRP3 [Figure 6e]. These findings denote that GP may exert its effects by inhibiting the TXNIP/NLRP3 pathway against CH-induced myocardial cell necrosis.
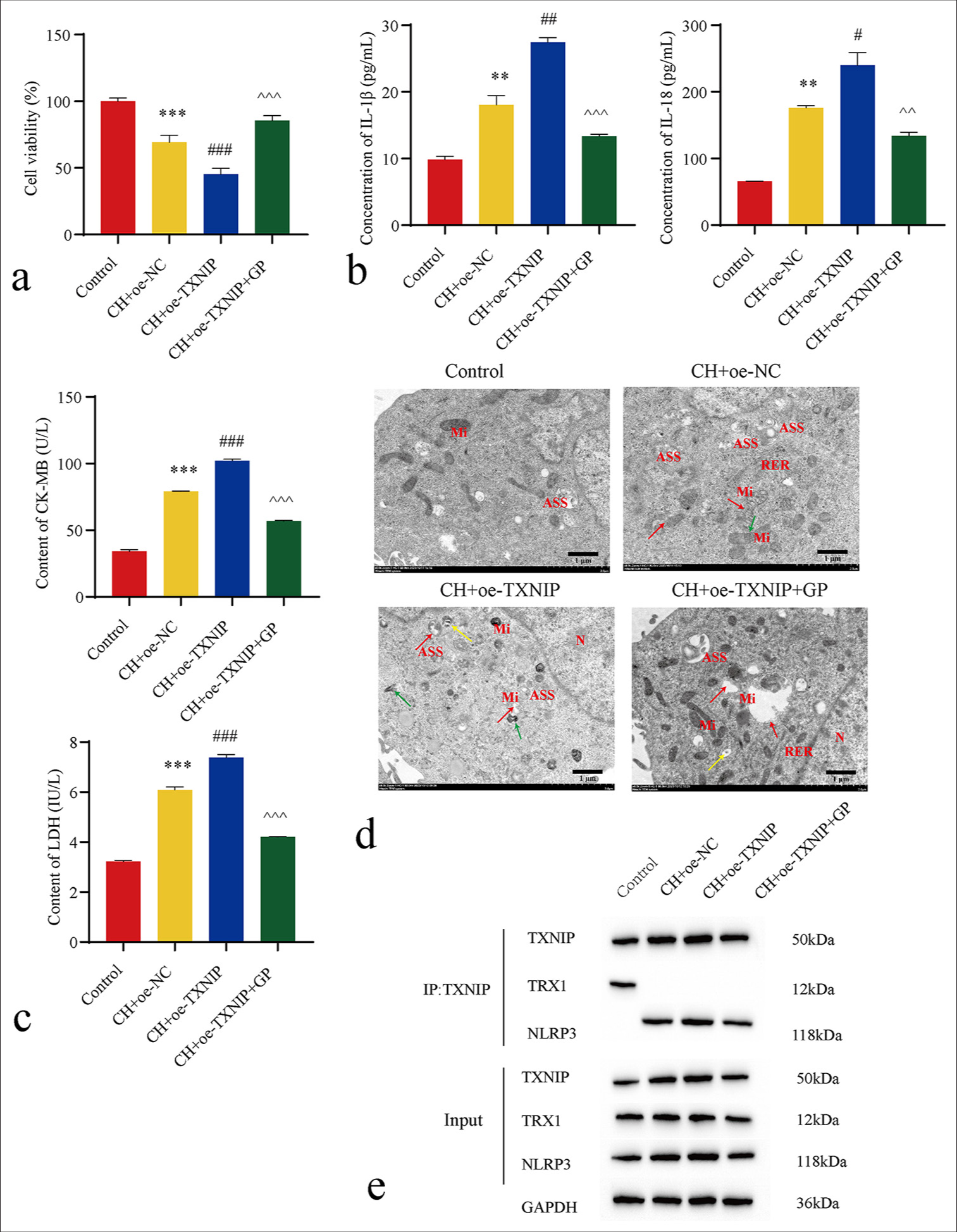
- Geniposide (GP) inhibits the thioredoxin-interacting protein (TXNIP)/NLRP3 signaling pathway to counter chronic hypoxia (CH)-Induced myocardial cell necrosis (n = 3). (a) Cell counting kit-8 detected cell viability; (b and c) enzyme-linked immunosorbent assay-detected interleukin (IL)-1β, IL-18, lactate dehydrogenase (LD), and creatine kinase-myocardial band (CK-MB) levels; (d) Transmission electron microscopy-detected cell pyroptosis. N: Nucleus, Mi: Mitochondria, ASS: Autophagolysosome, ERE: Rough endoplasmic reticulum, red arrow: Mitochondrial membrane rupture, green arrow: Mitochondrial deformation and shrinkage, yellow arrow: Mitochondrial vacuolation. (e) Immunoprecipitation-detected interaction between TXNIP and NLRP3. Compared with the control group, ✶✶P < 0.05, ✶✶✶P < 0.001; as compared with the CH + overexpression (oe)-NC group, #P < 0.05, ##P < 0.01, ###P < 0.001; as compared with the CH + oe-TXNIP group, ^^P < 0.01, ^^^P < 0.001. oe-NC: Overexpressing negative control, NLRP3: NOD-like receptor protein 3, TRX: Thioredoxin.
DISCUSSION
This work involved the examination of the involvement of the TXNIP/NLRP3 signaling pathway in pyroptosis triggered after MI and exploration of the potential effects of NLRP3 inhibitors and TXNIP overexpression on GP’s antipyroptosis properties of GP. Research findings indicate that GP treatment considerably enhanced cardiac structure and function. Throughout the treatment process, no notable changes were observed in the body weight or blood glucose levels of the mice, which suggests the negligible influence of GP on the overall metabolic status. Nevertheless, the treatment group exhibited heart rate regulation, possibly due to the direct influence of GP on the heart, which further confirms its role in cardiac function improvement. In vitro experiments further supported these findings. GP exerted significant protective effects on a CH-induced cardiomyocyte necrosis model. Furthermore, Ma et al. revealed GP’s protective effects against cardiac hypertrophy.[26] A mouse model of aortic constriction revealed that GP can inhibit cardiomyocyte hypertrophy, improve ventricular structure, and enhance cardiac function. Similarly, Li et al.[17] reported that in a transverse aortic constriction mouse model, GP greatly mitigated hypertrophic responses by increasing the left ventricular diastolic diameter and decreasing fractional values. Likewise, mice with aortic constriction showed a decline in cardiac function, with reduced EF and FS coupled with a notable increase in the left ventricular diastolic diameter.[27] GP treatment alleviated these morphological modifications. In summary, the above research findings suggest the potential of GP therapy in improving cardiac structure and function and its notable progress in providing protection against myocardial hypertrophy. Further analysis of cardiac tissue indicators revealed a significant increase in the relative heart weight of the MI group, which was likely due to cardiomyocyte loss and fibrous tissue proliferation. Conversely, the GP treatment group presented a substantial reduction in the infarcted area, which indicates that GP can effectively alleviate myocardial damage and prevent myocardial fibrosis progression. This protective effect may be achieved through the suppression of Col I and Col III gene expressions. The extracellular matrix of cardiac myocytes forms a complex protein network primarily composed of Col I and Col III.[28] These proteins contribute to the structural support of cardiac myocytes and are vital for the proper heart functioning. Research indicated a correlation between the excessive synthesis and insufficient degradation of Col I and Col III with cardiac fibrosis, which leads to their overaccumulation in myocardial tissues.[29] The balance between Col I and Col III synthesis and degradation notably affects myocardial remodeling and function in patients with diabetic heart failure.[30] Furthermore, GP exerted a protective effect on cardiac myocytes by reducing the pyroptosis levels and significantly decreasing the expressions of proteins, such as TXNIP, NLRP3, caspase-1, and GSDMD-N, in cardiac tissue. These molecules play pivotal roles in inflammation and cellular pyroptosis;[31,32] suppressing their expressions may be one of the mechanisms by which GP modulates post-MI pyroptosis. Rong et al.[33] demonstrated that GP significantly alleviated the cardiac myocyte damage caused by ischemia- RI in rat models, and this finding highlights its protective effects on the myocardium. Further investigations on the role of NLRP3 in GP-mediated antipyroptosis revealed that inhibition of NLRP3 also reduced the levels of NLRP3, TXNIP, caspase-1, and GSDMD-N in cardiac tissue while simultaneously decreasing ROS production. Excessive ROS within MI has a potential to induce damage to mitochondrial DNA (mtDNA), which leads to an imbalance amid enzymes of the respiratory chain complex and expressions of mtDNA-encoded genes. This condition, in turn, contributes to structural changes in the heart, which ultimately triggers the onset of heart failure.[34] Therefore, the NLRP3 signaling pathway is a critical participant in MI-induced pyroptosis.
Continued in vitro studies were conducted to explore the effect of GP on CH-induced cardiomyocyte pyroptosis and determine whether GP exerts its protective effects against CH-induced cardiomyocyte pyroptosis by inhibiting the TXNIP/NLRP3 signaling pathway. The research results demonstrate the substantial protection provided by GP against CH-induced cardiomyocyte pyroptosis. Compared with the control group, the GP treatment group showed no notable cytotoxicity. By contrast, the CH group showed a significant decrease in cardiomyocyte viability, accompanied with heightened pyroptosis rates and elevated levels of inflammatory markers IL-1β and IL-18. In addition, a significant increase was observed in the release of LDH and CK-MB in the CH group. Moreover, the study revealed that GP effectively reduced the myocardial cell pyroptosis induced by CH by inhibiting the TXNIP/NLRP3 signaling pathway. This finding was manifested by enhanced survival rates of cardiomyocytes, reduced rates of pyroptosis, and decreased release of inflammatory factors and markers of cellular damage. Yoshioka et al.[35] reported that in the TXNIP-knockout MI/RI mouse model, the recovery after ischemia/RI was better than that in the general MI/RI mice. This outcome implies that TXNIP is crucial in myocardial ischemia-RI, with GP potentially providing protective effects through TXNIP inhibition. Liu et al.[36] demonstrated that GP enhanced cellular resilience to endoplasmic reticulum stress and safeguarded pancreatic β-cells exposed to high glucose levels by facilitating the degradation of TXNIP protein. This result further supports the potential role of GP in TXNIP regulation and may play a role in other cell types. Fu et al.[37] suggested that GP decreased inflammatory cytokine levels by inhibiting NLRP3 inflammasome activation and boosted autophagy in brain vasculature-2 microglial cells after oxygen– glucose deprivation and reoxygenation. These findings indicate that GP not only protects cardiomyocytes but also suppresses the NLRP3 inflammasome in other cell types.
In summary, our findings indicate that GP, by modulating TXNIP/NLRP3 signaling pathway, enhances cardiac structure and function while reducing the extent of MI and the incidence of myocardial cell pyroptosis. This finding provides crucial groundwork for the further exploration of GP as a potential pharmaceutical intervention for post-heart attack complications. Nevertheless, this study had certain limitations, including the lack of comprehensive mechanistic investigations and clinical trial validation. Therefore, further in-depth research must be conducted to ascertain its prospects and safety profiles.
SUMMARY
GP drastically enhanced cardiac structure and function by modulating the TXNIP/NLRP3 signaling pathway, which concomitantly reduced the MI area and fibroblast proliferation. The inhibition of NLRP3 also lowered the levels of NLRP3, TXNIP, caspase-1, and GSDMD-N in cardiac tissues, which results in a decreased ROS production. This finding suggests that NLRP3 is a crucial player in GP’s antipyroptotic effects of GP, which potentially safeguards the myocardium by regulating mechanisms, such as cell pyroptosis, inflammatory responses, and oxidative stress. Moreover, after OE-TXNIP treatment, GP treatment significantly alleviated the cardiomyocyte necrosis caused by TXNIP overexpression, which further confirmed the effectiveness of GP in counteracting TXNIP-induced myocardial cell damage. Our discoveries establish a theoretical groundwork for future research and a formulation of therapeutic approaches aimed at TXNIP/NLRP3, as well as offering a potential approach for GP treatment in MI.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
ABBREVIATIONS
CCK-8: Cell Counting Kit-8
cDNA: Complementary deoxyribonucleic acid
CH: Chronic hypoxia
CK-MB: Creatine kinase-muscle/brain
Col I: Collagen I
Col III: Collagen III
EDV: End diastolic volume
EF: Ejection fraction
ELISA: Enzyme-Linked Immunosorbent Assay
ESV: End systolic volume
FS: Fractional shortening
GLP-1: Glucagon-like peptide-1
GP: Geniposide
GSDMD: Gasdermin D
HE: Hematoxylin-Eosin
IF: Immunofluorescence
IHC: Immunohistochemistry
IL-18: Interleukin-18
IL-1β: Interleukin-1β
IP: Immunoprecipitation
LDH: Lactate Dehydrogenase
LVIDd: Left ventricular internal diameter at diastole
LVIDs: Left ventricular internal diameter at systole
MCC950: NLRP3 inhibitor
MI: Myocardial infarction
MI/RI: Myocardial ischemia/reperfusion injury
mtDNA: Mitochondrial DNA
NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3
oe-NC: Overexpression-negative control
qPCR: Quantitative polymerase chain reaction
ROS: Reactive oxygen species
SPF: Specific pathogen free
TEM: Transmission electron microscopy
TRX: Thioredoxin
TTC: Triphenyltetrazolium Chloride
TUNEL: Terminal deoxynucleotidyl transferase 2’-Deoxyuridine 5’-triphosphate nick end labelling
TXNIP: Thioredoxin-interacting protein
WB: Western Blot
AUTHOR CONTRIBUTIONS
YQJ: Conceptualization, data curation, methodology, software, writing – original draft; YS: Conceptualization, methodology, software; CL, WWJ, and YW: Formal analysis; GLC: Validation; YL and HHH: Supervision, writing – review and editing.
ACKNOWLEDGMENT
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal procedures employed in the project was approved by Institutional Animal Care and Use of Chongqing Medical University (IACUC-CQMU), Approval number: IACUCCQMU-2023-0315, dated 2023.10.26. This article does not involve patients, so informed consent is not required.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This study was sponsored by Natural Science Foundation of Chongqing, China (Grant No. CSTB2022NSCQ-MSX1068).
References
- Characterization of dysregulated lncRNA-mRNA network based on ceRNA hypothesis to reveal the occurrence and recurrence of myocardial infarction. Cell Death Discov. 2018;4:35.
- [CrossRef] [PubMed] [Google Scholar]
- Septin4 promotes cardiomyocytes apoptosis by enhancing the VHL-mediated degradation of HIF-1α. Cell Death Discov. 2021;7:172.
- [CrossRef] [PubMed] [Google Scholar]
- The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725-30.
- [CrossRef] [PubMed] [Google Scholar]
- NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev. 2017;2017:9743280.
- [CrossRef] [PubMed] [Google Scholar]
- Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta. 2020;510:62-72.
- [CrossRef] [PubMed] [Google Scholar]
- GDF11 alleviates neointimal hyperplasia in a rat model of artery injury by regulating endothelial NLRP3 inflammasome activation and rapid re-endothelialization. J Transl Med. 2022;20:28.
- [CrossRef] [PubMed] [Google Scholar]
- Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5.
- [CrossRef] [PubMed] [Google Scholar]
- TRIM21 regulates pyroptotic cell death by promoting Gasdermin Doligomerization. Cell Death Differ. 2022;29:439-50.
- [CrossRef] [PubMed] [Google Scholar]
- The role of the NLRP3 inflammasome in mediating glomerular and tubular injury in diabetic nephropathy. Front Physiol. 2022;13:907504.
- [CrossRef] [PubMed] [Google Scholar]
- The emerging role of thioredoxin-interacting protein in myocardial ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2017;22:219-29.
- [CrossRef] [PubMed] [Google Scholar]
- A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221-5.
- [CrossRef] [PubMed] [Google Scholar]
- Ilexgenin A inhibits endoplasmic reticulum stress and ameliorates endothelial dysfunction via suppression of TXNIP/NLRP3 inflammasome activation in an AMPK dependent manner. Pharmacol Res. 2015;99:101-15.
- [CrossRef] [PubMed] [Google Scholar]
- Thioredoxin/txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514.
- [CrossRef] [PubMed] [Google Scholar]
- Pyroptosis-triggered pathogenesis: New insights on antiphospholipid syndrome. Front Immunol. 2023;14:1155222.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide from Gardenia jasminoides var. radicans Makino attenuates myocardial injury in spontaneously hypertensive rats via regulating apoptotic and energy metabolism signalling pathway. Drug Des Devel Ther. 2021;15:949-62.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide attenuates Aβ(25-35)-induced neurotoxicity via the TLR4/NF-κB pathway in HT22 cells. RSC Adv. 2018;8:18926-37.
- [CrossRef] [PubMed] [Google Scholar]
- Antioxidative property and molecular mechanisms underlying geniposide-mediated therapeutic effects in diabetes mellitus and cardiovascular disease. Oxid Med Cell Longev. 2019;2019:7480512.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide protects against acute alcohol-induced liver injury in mice via up-regulating the expression of the main antioxidant enzymes. Can J Physiol Pharmacol. 2015;93:261-7.
- [CrossRef] [PubMed] [Google Scholar]
- Anti-inflammatory evaluation of gardenia extract, geniposide and genipin. J Ethnopharmacol. 2006;103:496-500.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide prevents hypoxia/reoxygenation-induced apoptosis in H9c2 Cells: Improvement of mitochondrial dysfunction and activation of GLP-1R and the PI3K/AKT signaling pathway. Cell Physiol Biochem. 2016;39:407-21.
- [CrossRef] [PubMed] [Google Scholar]
- Antithrombotic activities of aqueous extract from Gardenia jasminoides and its main constituent. Pharm Biol. 2013;51:221-5.
- [CrossRef] [PubMed] [Google Scholar]
- Eucommia ulmoides oliver extract, aucubin, and geniposide enhance lysosomal activity to regulate ER stress and hepatic lipid accumulation. PLoS One. 2013;8:e81349.
- [CrossRef] [PubMed] [Google Scholar]
- Isolation of antifungal compounds from Gardenia jasminoides. Pak J Biol Sci. 2009;12:949-56.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of autophagy by geniposide protects against myocardial ischemia/reperfusion injury. Int Immunopharmacol. 2020;85:106609.
- [CrossRef] [PubMed] [Google Scholar]
- Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271-96.
- [CrossRef] [PubMed] [Google Scholar]
- Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor/AMPKα signalling pathway. Br J Pharmacol. 2016;173:1502-16.
- [CrossRef] [PubMed] [Google Scholar]
- A review on the phytochemistry, pharmacology, pharmacokinetics and toxicology of geniposide, a natural product. Molecules. 2017;22:1689.
- [CrossRef] [PubMed] [Google Scholar]
- Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation. 1999;99:2750-6.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac interstitium in health and disease: The fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637-52.
- [CrossRef] [PubMed] [Google Scholar]
- Type 2 diabetes mellitus and chronic heart failure with midrange and preserved ejection fraction: A focus on serum biomarkers of fibrosis. J Diabetes Res. 2020;2020:6976153.
- [CrossRef] [PubMed] [Google Scholar]
- Thioredoxin-interacting protein promotes activation and inflammation of monocytes with DNA demethylation in coronary artery disease. J Cell Mol Med. 2020;24:3560-71.
- [CrossRef] [PubMed] [Google Scholar]
- HuNoV non-structural protein P22 induces maturation of IL-1β and IL-18 and N-GSDMD-dependent pyroptosis through activating NLRP3 inflammasome. Vaccines (Basel). 2023;11:993.
- [CrossRef] [PubMed] [Google Scholar]
- Protective effects of geniposide on hepatic ischemia/reperfusion injury. Transplant Proc. 2017;49:1455-60.
- [CrossRef] [PubMed] [Google Scholar]
- Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457-64.
- [CrossRef] [PubMed] [Google Scholar]
- Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122:267-79.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide accelerates proteasome degradation of Txnip to inhibit insulin secretion in pancreatic β-cells. J Endocrinol Invest. 2017;40:505-12.
- [CrossRef] [PubMed] [Google Scholar]
- Geniposide inhibits NLRP3 inflammasome activation via autophagy in BV-2 microglial cells exposed to oxygen-glucose deprivation/reoxygenation. Int Immunopharmacol. 2020;84:106547.
- [CrossRef] [PubMed] [Google Scholar]




