


Translate this page into:
Mitochondrial autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3-mediated pyroptosis and alleviates endothelial cell injury in pregnancy-induced hypertension

*Corresponding author: Xiangzhen Zhang, Department of Obstetrics, The Second People’s Hospital of Yueqing, Wenzhou, China. zxz53558@163.com
-
Received: ,
Accepted: ,
How to cite this article: Zhang M, Zhang X, Mao L. Mitochondrial autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3-mediated pyroptosis and alleviates endothelial cell injury in pregnancy-induced hypertension. CytoJournal. 2025;22:60. doi: 10.25259/Cytojournal_52_2025
Abstract
Objective:
Pregnancy-induced hypertension (PIH) is a common complication during pregnancy and is closely associated with vascular endothelial cell damage and nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3)-mediated pyroptosis. This study aimed to investigate whether mitophagy alleviates vascular endothelial cell damage in PIH by inhibiting NLRP3-mediated pyroptosis. The regulatory mechanisms of pyroptosis-related pathways were systematically investigated by establishing a cellular model of PIH and incorporating mitophagy intervention.
Material and Methods:
An Nω-nitro-L-arginine methyl ester (L-NAME)-induced gestational hypertension model was established, and the cell samples were grouped as follows: Control group (Control), L-NAME-induced gestational hypertension group (L-NAME), mitochondrial autophagy inhibition group (L-NAME+ 3-methyladenine [3-MA]), and mitochondrial autophagy activation group (L-NAME+ rapamycin [Rapa]). Cell viability was assessed through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, lactate dehydrogenase (LDH) levels were measured to evaluate cell damage, and reactive oxygen species (ROS) kits were used to quantify ROS accumulation. Cell death was evaluated using terminal deoxynucleotidyl transferase dUTP nick end labeling staining to detect apoptotic cells. Immunofluorescence, Western blot analysis, and quantitative real-time polymerase chain reaction were performed to assess the expression levels of proteins and genes associated with mitophagy (e.g., microtubule-associated protein 1 light chain 3 and sequestosome 1) and those linked to pyroptosis (e.g., NLRP3, gasdermin D (GSDMD), cysteinyl aspartate-specific proteinase 1 (caspase-1), interleukin (IL)-1β, and IL-18). The role of NLRP3 in pyroptosis regulation through mitochondrial autophagy was further examined using NLRP3 small interfering RNA (siNLRP3) transfection experiments.
Results:
L-NAME treatment substantially decreased vascular endothelial cell viability, elevated LDH release and ROS levels, and upregulated pyroptosis-related proteins (NLRP3, GSDMD, and caspase-1) and inflammatory factors (IL-1β and IL-18). The inhibition of mitochondrial autophagy with 3-MA further enhanced pyroptosis and aggravated cell damage, and its activation with Rapa reduced pyroptosis, improved cell survival, and decreased LDH release and ROS levels. NLRP3 silencing (siNLRP3) significantly inhibited pyroptosis and alleviated the cell damage caused by 3-MA. Meanwhile, Rapa enhanced the protective effect of NLRP3 silencing.
Conclusion:
This study demonstrates that mitophagy can effectively alleviate the vascular endothelial cell damage associated with PIH by inhibiting NLRP3-mediated pyroptosis. The findings provide new theoretical support for the treatment of PIH and suggest potential intervention targets.
Keywords
Mitophagy
Nucleotide-binding oligomerization domain-like receptor protein 3
Pregnancy-induced hypertension
Pyroptosis
Vascular endothelial cell injury

INTRODUCTION
Pregnancy-induced hypertension (PIH) is a pregnancy-specific condition characterized by increased blood pressure, proteinuria, and edema and often results in damage to multiple organs, thus posing a significant risk to maternal and fetal health. PIH remains a major contributor to maternal and perinatal mortality.[1,2] Hypertension leads to vascular endothelial damage.[3] Endothelial injury is associated with decreased nitric oxide availability, which ultimately initiates and exacerbates various vascular diseases.[4,5] The nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome is a protein complex in the NOD-like receptor family that features a pyrin domain and a heat shock protein domain and is closely associated with the onset of inflammatory disorders. When the NLRP3 inflammasome is activated, cysteinyl aspartate-specific proteinase 1 (caspase-1), a cysteinyl aspartate-specific protease, cleaves pro-interleukin-1β (IL-1β), pro-interleukin-18 (IL-18); and gasdermin D (GSDMD) to induce the maturation of IL-1β, IL-18, and the N-terminal fragment of GSDMD (GSDMD-N). This sequence of events initiates a specific form of inflammatory cell death known as pyroptosis.[6-8] During NLRP3 activation, damaged endothelial cells secrete various inflammatory mediators.[9] Furthermore, sustained NLRP3 activation accelerates pyroptosis, resulting in significant endothelial cell loss and subsequent endothelial dysfunction.[10] Inhibiting pyroptosis in endothelial cells enhances their survival, promotes angiogenesis, and improves the prognosis of ischemic diseases.[11] Thus, blocking NLRP3 activation may provide a promising approach to manage inflammatory conditions associated with pyroptosis.
NLRP3 activators, such as adenosine 5'-triphosphate (ATP) or streptolysin, induce mitochondrial dysfunction and lead to NLRP3 activation by decreasing mitochondrial membrane potential (MMP) and releasing mitochondrial damage-associated molecular patterns, including mitochondrial reactive oxygen species (ROS).[12-14] Therefore, mitochondrial dysfunction is crucial in triggering NLRP3 activation through multiple pathways.[15,16] The mitochondrial ROS scavenger mito-TEMPO can inhibit NLRP3 activation in cells,[17] suggesting a direct link between the oxidative stress from mitochondrial dysfunction and the activation of NLRP3 inflammasome during inflammation. Mitochondrial autophagy has gained increasing attention for its role as a critical cellular self-repair mechanism in various diseases.[18,19] It facilitates the removal of damaged mitochondria, making it crucial in maintaining mitochondrial balance and ensuring mitochondrial quality control.[20]
The binding of pro-IL-1α to mitochondrial cardiolipin inhibits the interaction between microtubule-associated protein 1 light chain 3B and mitochondria, preventing mitochondrial autophagy and leading to the accumulation of damaged mitochondria. This accumulation further stimulates NLRP3 activation and exacerbates IL-1β release.[21] Therefore, the pathological mechanisms of mitochondrial dysfunction, oxidative stress, and impaired mitochondrial autophagy collectively accelerate NLRP3 activation. Maintaining mitochondrial homeostasis and enhancing mitochondrial autophagy to suppress NLRP3 activation and subsequent pyroptosis may provide a viable strategy for treating inflammatory diseases.
The aim of this study is to investigate the role of mitophagy in mitigating vascular endothelial cell damage in PIH. Specifically, we seek to explore how mitophagy may influence the suppression of NLRP3 inflammasome activation and its subsequent impact on pyroptosis in endothelial cells. By elucidating the mechanisms through which mitophagy regulates mitochondrial homeostasis, we aim to provide deeper insights into its potential therapeutic effects on vascular injury in PIH.
MATERIAL AND METHODS
Cell culture
A human umbilical vein endothelial cell (HUVEC) line (PCS-100-010) was obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were authenticated through short tandem repeat profiling, and mycoplasma testing was conducted to prevent contamination. The cells were cultured individually in their respective culture media. The HUVECs were maintained in a HUVEC complete medium (10% fetal bovine serum, 100 U/mL penicillin-streptomycin, 1% endothelial cell culture supplement) (iCellh110-001b, iCell-h110, Shanghai, China), seeded into 25 cm2 culture flasks, and incubated in a 37°C, 5% carbon dioxide (CO2) incubator (BPN-80CH, Shanghai, China). Subculture was performed every 3–4 days based on cell growth, and cells were selected for experimental studies as necessary. The experimental groups were as follows: Control group, L-NAME group (PIH group, induced with 300 µM L-NAME for 24 h following the method of Chen et al.),[22] L-NAME + 3-methyladenine (3-MA; HY-19312, MedChemExpress, New Jersey, USA) group (hypertension group treated with 5 mM 3-MA for 2 h to inhibit mitochondrial autophagy), and L-NAME + rapamycin (Rapa; HY-10219, MedChemExpress, New Jersey, USA) group (hypertension group treated with 100 nM Rapa for 4 h to activate mitophagy).
Cell viability assay
Cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The treated HUVECs were seeded in 96-well plates (5 × 104 well/mL) and cultured until they reached the required confluence, after which they were treated under the respective conditions.
After treatments, the cells were added with the MTT (G4101, Servicebio, Wuhan, China) reagent and incubated for 4 h. Dimethyl sulfoxide (ST1276, Beyotime, Shanghai, China) was subsequently incorporated to dissolve the formed purple-blue formazan crystals. Absorbance at 490 nm was measured using a multifunctional microplate reader (CMaxplus, MOLECULAR DEVICES, USA) to determine cell viability. Each condition was evaluated in triplicate, and the experiment was performed at least 3 times.
Lactate dehydrogenase (LDH) assay
Following treatment as per the experimental groups, the cell supernatants were collected and centrifuged at 3,000 rpm for 20 min at 4°C. The concentration of LDH (A020-2, Nanjing Jiancheng, Nanjing, China) in the supernatant was quantified in accordance with the guidelines provided by the manufacturer of the enzyme-linked immunosorbent assay kit.
ROS assay
The treated cells were collected, and the culture medium was discarded. Then, 500 µL of diluted 10 µM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) (S0033S-1, Beyotime, Shanghai, China) solution was added to ensure full cell exposure. The cells were incubated at 37°C in an atmosphere containing 5% CO2 for 20 min. After incubation, the cells were thoroughly rinsed with serum-free culture medium 3 times to remove any unabsorbed DCFH-DA. The cells were then observed and photographed under a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan). Analysis and quantification were performed using ImageJ software (V1.46; National Institutes of Health, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
The cells were seeded into 24-well plates (5 × 104 well/mL) and cultured overnight, followed by apoptosis induction and two washes with phosphate-buffered saline (PBS) (G4202, Servicebio, Wuhan, China). The cells were then fixed with 4% paraformaldehyde at 4°C for 25 min, followed by two washes with PBS. The cells were permeabilized with a 0.2% Triton X-100 (T8200, Solarbio, Beijing, China) solution. After three washes with PBS, the cells were added with the labeling solution (K1133, APExBIO, Shanghai, China) and incubated in the dark for 60 min, followed by additional washes with PBS. The cells were stained with 4',6-diamidino-2-phenylindole (DAPI) (C0060, Solarbio, Beijing, China) and washed 3 times with PBS. An anti-fade reagent (S2100, Solarbio, Beijing, China) was added, and images were captured using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan).
MMP detection
Staining buffer and working solutions were prepared according to the instructions of the 5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) assay kit (C2006, Beyotime, Shanghai, China). In brief, a six-well plate was added with 1 mL of working solution each well, incubated for 20 min, and then observed and photographed using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan).
Immunofluorescence
The cells were cultured on coverslips placed in six-well plates (5 × 103 well/mL), fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% goat serum. Primary antibodies against microtubule-associated protein 1 light chain 3 (LC3) (14600-1-AP, Proteintech, Wuhan, China) and GSDMD (HA601046, HUABIO, Hangzhou, China) were applied, and the cells were incubated overnight at 4°C, followed by washing and incubation with the appropriate secondary antibodies (GB22303, Servicebio, Wuhan, China). After additional washes, the cells were stained with DAPI for 10 min. Finally, fluorescence images were captured using a fluorescence microscope (CKX53, OLYMPUS, Tokyo, Japan). Image analysis and quantification were performed using ImageJ software (V1.46, National Institutes of Health, USA).
Western blot (WB) analysis
Following treatment, the cells were washed 3 times with PBS and lysed on ice with radio immunoprecipitation assay buffer (P0013C, Beyotime, Shanghai, China) for 30 min to extract total protein. The proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (P0015A, Beyotime, Shanghai, China) and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked for 2 h and then incubated overnight at 4°C with the primary antibodies against LC3 (1:1,000 dilution, 14600-1-AP, Proteintech, Wuhan, China), sequestosome 1 (P62) (1:1,000 dilution, 80294-1-RR, Proteintech, Wuhan, China), NLRP3 (1:1,000 dilution, 68102-1-lg, Proteintech, Wuhan, China), caspase-1 (1:1,000 dilution, ET1608-69, HUABIO, Hangzhou, China), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000 dilution, TA-08, ZSGB-BIO, Beijing, China). The membrane was then incubated with the appropriate secondary antibodies (1:2000 dilution, ZB-2305, ZSGB-BIO, Beijing, China) at room temperature for 2 h. Chemiluminescence detection was performed using the enhanced chemiluminescence reagent (Tanon-4600, Yuanping Hao Biotechnology, Beijing, China), and images were acquired using a gel imaging system. Protein band intensities were quantified using ImageJ software (V1.46, National Institutes of Health, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
After treatment, total RNA was extracted from the cells using the TRNzol Universal RNA Extraction Reagent (DP424, TIANGEN Biotech, Beijing, China). The extracted RNA was subsequently reverse transcribed into complementary DNA using the reverse transcription kit (KR116, TIANGEN, Beijing, China). PCR amplification was performed using specific forward and reverse primers on a real-time fluorescence quantitative PCR system (LightCycler96, Roche, Switzerland), with GAPDH as the internal control.[22] The relative gene expression levels were quantified using the 2−△△CT method. The sequences of the primers are provided in Table 1.
| Gene | Primer sequences (5'-3') |
|---|---|
| LC3 | R: GCTCGTAGATGTCCGCGAT F: AACATGAGCGAGTTGGTCAAG |
| P62 | R: AGTGTCCGTGTTTCACCTTCC F: GACTACGACTTGTGTAGCGTC |
| NLRP3 | R: ATCTGAAGCTCTGGTTGGTC F: GAGTCTCCCAAGGCATTCTCC |
| GSDMD | R: TGGTGTTGTCCTCCGGAATG F: GCGTTTCACTCAGCATGGTC |
| Caspase-1 | R: GTTCCATGGGTGAAGATAATGTTT F: TGAAAATCGAACCTTGCGGA |
| IL-1β | R: TTCAACACGCAGGACAGGTACAG F: CCAGGGACAGGATATGGAGCA |
| IL-18 | R: CTGCCACCTGCTGCAGTCTA F: TCTACTGGTTCAGCAGCAGCCATCTTTA |
| GAPDH | R: AAGGTCGGAGTCAACGGATTT F: TTCTCAGCCTTGACGGTGC |
LC3: Microtubule-associated protein 1 light chain 3, P62: Sequestosome 1, NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3, GSDMD: Gasdermin-D, Caspase-1: Cysteine-aspartic acid protease-1, IL-1β: Interleukin-1β, IL-18: Interleukin-18, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
NLRP3 silencing experiment
The small interfering RNA (siRNA) targeting the NLRP3 gene (siNLRP3) and the control siRNA (nontargeting control [siNC]) used for transfection were designed and synthesized by Jiangsu Kaiji Biotechnology Co., Ltd. (China). The siNLRP3 sequences were 5'-GUAUGAGAGUAGAGCGAUUUU-3' and 5'-GCUAAUGAUCGACUUCAAU-3', and the siNC (negative control) sequences were 5'-UUCUCCGAACGUGUCACGUTT-3' and 5'-ACGUGACACGUUCGGAGAATT-3'. HUVECs in the logarithmic growth phase were seeded in six-well plates and cultured until they reached approximately 80% confluence. The cells were then washed with PBS once and transferred to a serum-free medium. In accordance with the manufacturer’s protocol for Lipofectamine 2000 (11668027, Thermo Fisher Scientific, Waltham, MA, USA), the siRNA was mixed with the transfection reagent and applied to the cells, which were then incubated for 48 h. After incubation, the transfected cells were harvested and assigned to different experimental groups as follows: L-NAME + siNC (siNC), L-NAME + siNLRP3 (siNLRP3), L-NAME + siNLRP3 + 3-MA (siNLRP3 + 3-MA), and L-NAME + siNLRP3 + Rapa (siNLRP3 + Rapa). Subsequent experiments were performed as previously described.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism software (version 9.5, GraphPad Software, La Jolla, CA, USA). Data for normally distributed quantitative variables were expressed as the mean ± standard deviation. Comparisons among multiple groups were performed using one-way analysis of variance, followed by Bonferroni’s post hoc test. Pairwise comparisons were conducted using an independent sample t-test. Comparisons between two groups were analyzed using the t-test. Post hoc analysis was performed using the Tukey method. A P < 0.05 was considered statistically significant.
RESULTS
Effect of mitophagy on the viability of HUVECs and the expression of autophagy-related proteins
The role of mitophagy in L-NAME-induced vascular endothelial cell injury was investigated, and the following experimental results were obtained:
Figure 1a shows that exposure to 300 µM L-NAME for 24 h resulted in a significant decrease in cell viability. MTT assay indicated that L-NAME significantly inhibited cell activity, and 3-MA further exacerbated this effect. Meanwhile, Rapa significantly enhanced cell viability compared with L-NAME alone [Figure 1b] (P < 0.01). As shown in Figure 1c and d, immunofluorescence analysis of LC3II/LC3I expression demonstrated that L-NAME markedly reduced fluorescence intensity, which was further exacerbated by 3-MA. By contrast, Rapa significantly increased fluorescence intensity. WB analysis of LC3II/LC3I and P62 protein expression [Figure 1e-g] revealed that L-NAME significantly downregulated LC3II/LC3I while upregulating P62, and 3-MA further exacerbated these effects. By contrast, Rapa significantly upregulated LC3II/LC3I (P < 0.01) and downregulated P62 (P < 0.01) [Figure 1e-g]. These findings were consistent with the immunofluorescence results, as similar trends were observed for LC3 and P62 expression at the messenger RNA level [Figure 1h and i]. In conclusion, mitophagy plays a crucial protective role in L-NAME-induced vascular endothelial cell injury.
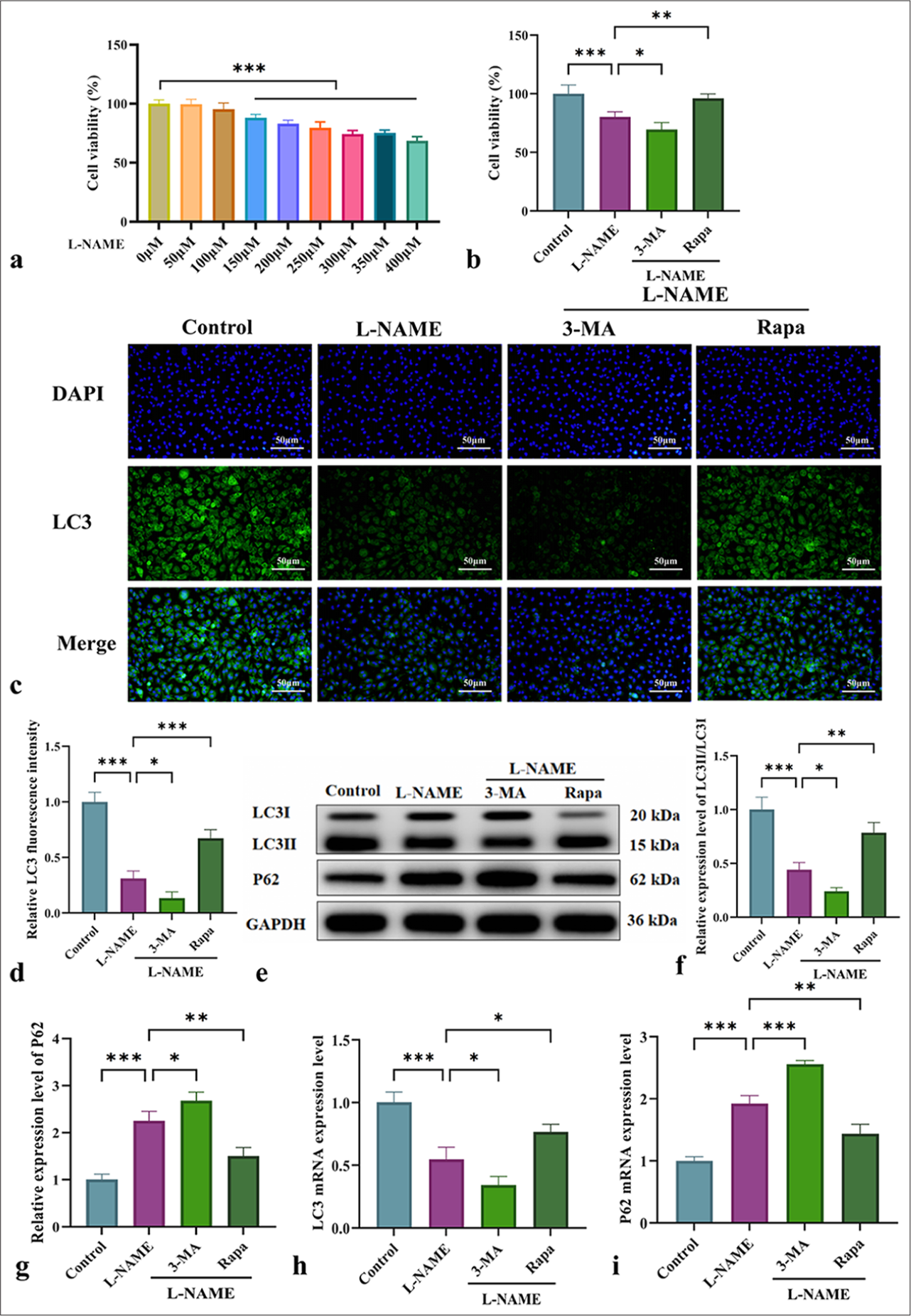
- Effects of mitophagy on cell viability and autophagy-related protein expression in HUVECs. (a) Effects of different concentrations (0–400 µM) of L-NAME on the 24-h viability of HUVECs; the horizontal line in the upper right corner of the bar graph indicates that the cell viability under L-NAME concentrations of 150–400 µM is significantly different from that at 0 µM (P < 0.001). (b) Cell viability of HUVECs in different groups. (c) Immunofluorescence analysis of LC3II/LC3I expression in HUVECs from various groups (at ×200 magnification, scale bar: 50 µm). (d) LC3 immunofluorescence quantification graph. (e) Protein levels of LC3II/LC3I and P62 detected by WB. (f and g) Quantitative analysis of autophagy-related proteins LC3II/LC3I and P62. (h and i) Messenger RNA levels of LC3 and P62 detected by qRT-PCR. n = 3, Data are shown as the means ± SD. (✶P < 0.05, ✶✶P < 0.01 ✶✶✶P < 0.001, vs. L-NAME, NS indicates no significance). HUVECs: Human umbilical vein endothelial cells, L-NAME: Nω-Nitro-L-arginine methyl ester, 3-MA: 3-Methyladenine, RaPa: Rapamycin, LC3: Microtubule-associated protein 1 light chain 3, DAPI: 4',6-diamidino-2-phenylindole, LC3II: Microtubule-associated protein 1 light chain 3 type II, P62: Sequestosome 1, LC3I: Microtubule-associated protein 1 light chain 3 type I, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, qRT-PCR: Quantitative real-time polymerase chain reaction, WB: Western blot, SD: Standard deviation.
Effect of mitophagy regulation on HUVEC damage and oxidative stress
The effects of different treatments on cell damage and oxidative stress were compared to investigate the role of mitophagy in L-NAME-induced endothelial cell injury.
TUNEL staining [Figure 2a and b] revealed a significant increase in fluorescence intensity after L-NAME treatment, indicating cell damage. This increase was further enhanced by 3-MA but significantly diminished by Rapa (P < 0.001). Measurement of LDH levels in HUVECs across different groups revealed that L-NAME significantly elevated the LDH levels, indicating cellular damage. 3-MA further increased the LDH levels, and Rapa significantly decreased them [Figure 2c] (P < 0.05). This finding was consistent with the TUNEL staining results. ROS fluorescence analysis was performed to assess the effect of L-NAME-induced oxidative stress. Figure 2d and e shows that L-NAME markedly elevated the ROS levels, indicating that it induces oxidative stress and endothelial cell damage. 3-MA further elevated the ROS levels, and Rapa significantly reduced them (P < 0.01). These findings suggested that mitophagy exerts a protective effect against L-NAME-induced endothelial cell damage by modulating oxidative stress and regulating cell death pathways.

- Influence of mitophagy regulation on HUVEC damage and oxidative stress. (a) Microscopic images of TUNEL staining in HUVECs from each group (at ×200 magnification, scale bar: 50 µm). (b) Statistical analysis of the number of cells stained by TUNEL. (c) LDH release in HUVECs from different groups. (d) ROS expression in HUVECs from each group (at ×200 magnification, scale bar: 50 µm). (e) Bar graph statistics of ROS levels. (f) JC-1 staining in HUVECs of each group (at ×100 magnification, scale bar: 100 µm). (g) JC-1 fluorescence quantification graph. n = 3, Data are shown as the means ± SD. (✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001, vs. L-NAME, NS indicates no significance). HUVECs: Human umbilical vein endothelial cells, L-NAME: Nω-Nitro-L-arginine methyl ester, 3-MA: 3-Methyladenine, Rapa: Rapamycin, DAPI: 4',6-diamidino-2-phenylindole, TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling, LDH: Lactate dehydrogenase, ROS: Reactive oxygen species, JC-1: 5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide, SD: Standard deviation.
JC-1 dye staining was performed to assess MMP in HUVECs to further investigate the protective role of mitophagy in cellular injury [Figure 2f and g]. The control group displayed a high MMP, characterized by strong red fluorescence. After L-NAME treatment, the MMP decreased, indicating mitochondrial dysfunction. 3-MA exacerbated the damage, and Rapa promoted mitophagy, restored red fluorescence, and alleviated L-NAME-induced injury. These results indicated that mitophagy plays a pivotal role in endothelial cell injury associated with gestational hypertension. Its activation can suppress NLRP3-mediated pyroptosis, thereby mitigating cellular damage.
Effect of mitophagy on NLRP3-mediated pyroptosis and inflammatory marker regulation
The involvement of NLRP3-induced pyroptosis in endothelial cell damage following L-NAME treatment was investigated by evaluating the effects of different interventions on the expression of NLRP3, GSDMD, caspase-1, and pro-inflammatory factors (IL-1β and IL-18). Immunofluorescence, WB, and qRT-PCR analyses [Figure 3] demonstrated that L-NAME significantly activated the NLRP3 inflammasome, enhanced GSDMD cleavage, and promoted caspase-1 activation, thus initiating pyroptosis and the secretion of inflammatory mediators such as IL-1β and IL-18 [Figure 3]. Following 3-MA intervention, pyroptosis was further increased, accompanied by enhancements in NLRP3 inflammasome activation and GSDMD cleavage [Figure 3]. Conversely, Rapa alleviated NLRP3-mediated pyroptosis by activating mitophagy, thus significantly reducing the inflammatory response [Figure 3] (P < 0.05 or P < 0.001). In summary, mitophagy may serve as a potential therapeutic target for mitigating vascular endothelial damage associated with conditions such as gestational hypertension by modulating pyroptosis and NLRP3 inflammasome activation.
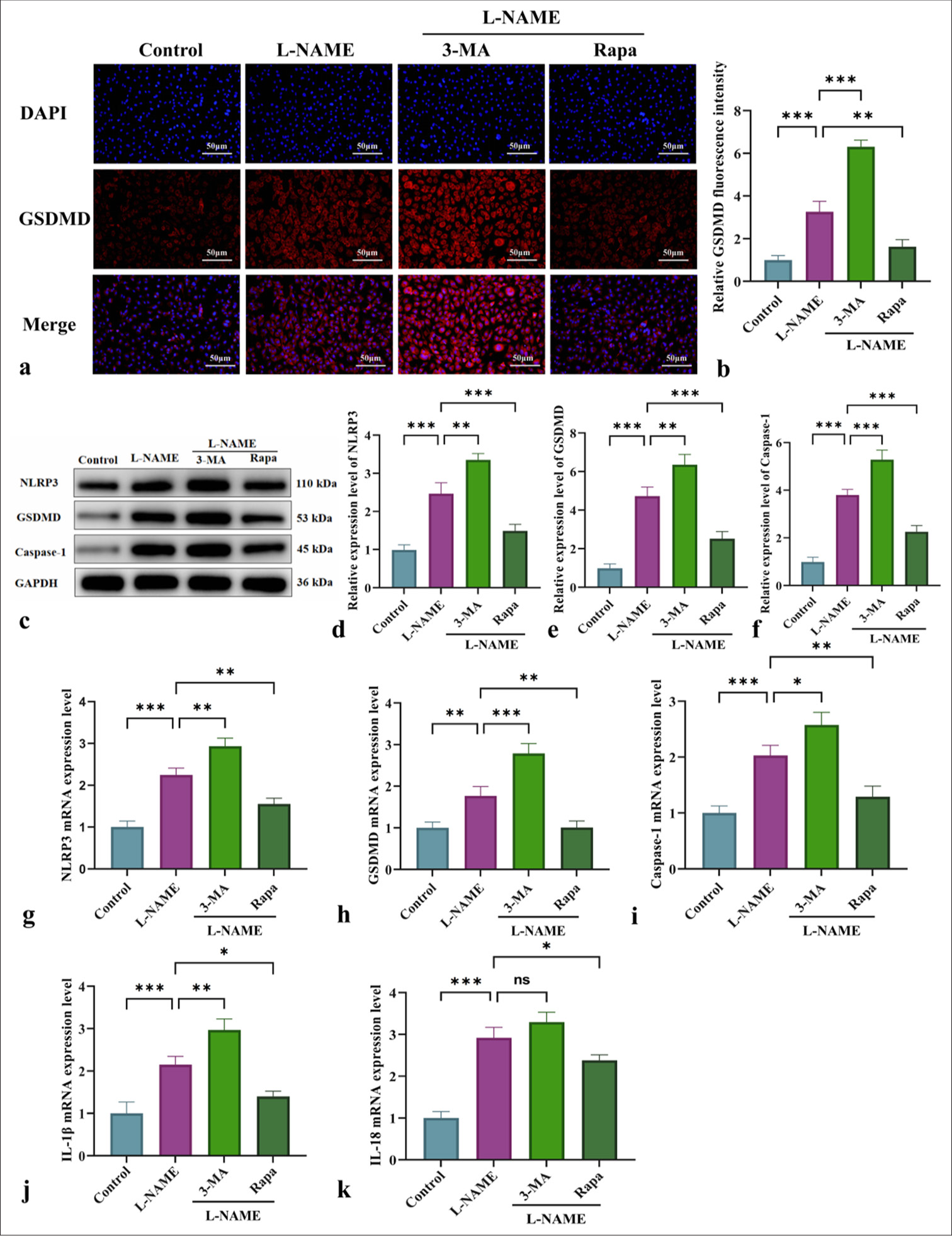
- Effect of mitophagy on the modulation of NLRP3-driven pyroptosis and inflammation-associated markers. (a) Immunofluorescent detection of GSDMD expression in HUVECs from different experimental groups (at ×200 magnification, scale bar: 50 µm). (b) GSDMD immunofluorescence quantification graph. (c) WB for the detection of NLRP3, GSDMD, and caspase-1 protein levels. (d-f) Quantitative analysis of the content of pyroptosis-related proteins NLRP3, GSDMD, and caspase-1. (g-k) qRT-PCR for the measurement of NLRP3, GSDMD, caspase-1, IL-1β, and IL-18 messenger RNA expression levels. n = 3, Data are shown as the means ± SD. (✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001, vs. L-NAME, NS indicates no significance). HUVECs: Human umbilical vein endothelial cells, L-NAME: Nω-Nitro-L-arginine methyl ester, 3-MA: 3-Methyladenine, Rapa: Rapamycin, NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3, GSDMD: Gasdermin-D, IL-1β: Interleukin-1β, DAPI: 4',6-diamidino-2-phenylindole, Caspase-1: Cysteine-aspartic acid protease-1, IL-18: Interleukin-18, WB: Western blot, qRT-PCR: Quantitative real-time polymerase chain reaction, SD: Standard deviation.
Effect of NLRP3 silencing combined with mitophagy regulation on the cell viability and autophagy levels of L-NAME-treated HUVECs
The impact of NLRP3 silencing on the cell viability and autophagy levels of L-NAME-treated HUVECs was assessed by interfering with NLRP3 expression and using mitochondrial autophagy-related drugs 3-MA and Rapa. MTT assay results in Figure 4a demonstrated that NLRP3 silencing reduced cell viability following L-NAME treatment, with a further decline observed after 3-MA intervention. Conversely, Rapa significantly enhanced cell viability. Immunofluorescence analysis revealed a significant increase in LC3II/LC3I fluorescence intensity in the NLRP3-silenced (siNLRP3) group. However, autophagy activation was suppressed by 3-MA, resulting in a reduction in fluorescence intensity. By contrast, Rapa markedly increased autophagy fluorescence intensity [Figure 4b and c]. WB and qRT-PCR analyses further confirmed the trends in LC3II/LC3I and P62 expression patterns, indicating that silencing NLRP3 improved L-NAME-induced cell damage by regulating mitophagy [Figure 4d-h]. The most significant effect was observed with Rapa intervention [Figure 4d-h] (P < 0.01). In summary, mitophagy plays a critical protective role in mitigating NLRP3-mediated cell damage.
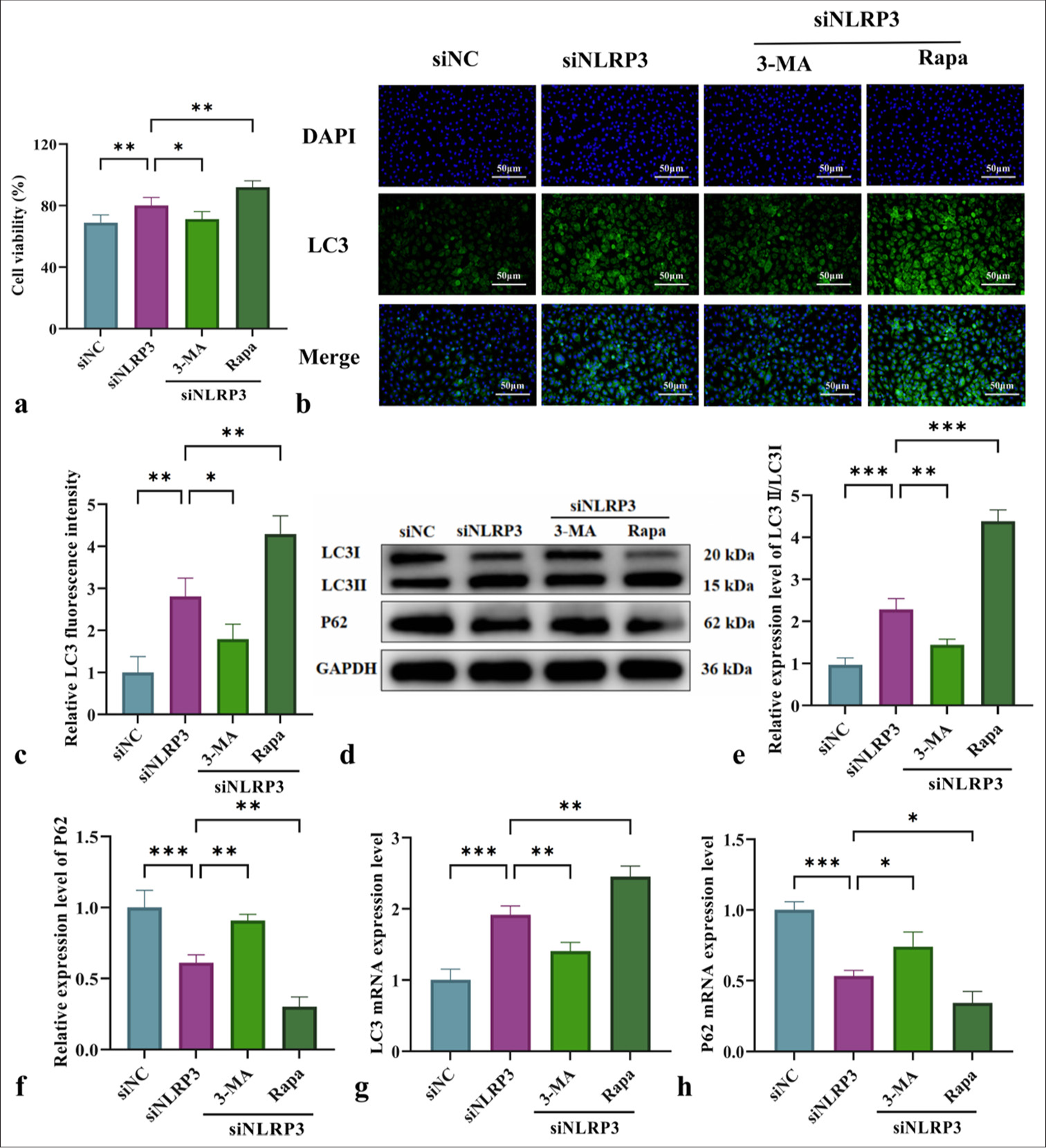
- Impact of NLRP3 silencing combined with mitochondrial autophagy regulation on the cell viability and autophagy levels of L-NAME-treated HUVECs. (a) Effects of L-NAME on the 24-h survival rate of HUVECs in different groups. (b) Immunofluorescence analysis of LC3II/LC3I expression in HUVECs from various groups (at ×200 magnification, scale bar: 50 µm). (c) LC3 immunofluorescence quantification graph. (d) WB for the detection of LC3II/LC3I and P62 protein levels. (e and f) Quantitative analysis of the content of autophagy-related proteins LC3II/LC3I and P62. (g and h) qRT-PCR for the detection of LC3 and P62 messenger levels. n = 3, Data are shown as means ± SD. (✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001, vs. siNLRP3, NS indicates no significance). NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3, L-NAME: Nω-Nitro-L-arginine methyl ester, HUVECs: Human umbilical vein endothelial cells, 3-MA: 3-Methyladenine, Rapa: Rapamycin, LC3: Microtubule-associated protein 1 light chain 3, DAPI: 4',6-diamidino-2-phenylindole, LC3II: Microtubule-associated protein 1 light chain 3 type II, GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, LC3I: Microtubule-associated protein 1 light chain 3 type I, P62: Sequestosome 1, WB: Western blot, qRTPCR: Quantitative real-time polymerase chain reaction, SD: Standard deviation.
Effect of NLRP3 silencing combined with mitophagy regulation on L-NAME-induced cell damage and oxidative stress in HUVECs
The role of NLRP3 silencing and mitochondrial autophagy regulation on L-NAME-induced vascular endothelial cell injury was assessed across different treatment groups. As shown in Figure 5, L-NAME significantly increased the LDH levels, ROS production, and number of TUNEL-positive cells, indicating aggravated cellular injury. Figure 5 demonstrates that NLRP3 silencing (siNLRP3 group) partially alleviated these injuries. Further modulation of mitochondrial autophagy, either through inhibition by 3-MA or activation by Rapa, exerted differential regulatory effects on cellular injury. 3-MA aggravated cell damage, and Rapa effectively alleviated these injuries, showing a significant regulatory effect [Figure 5a-e] (P < 0.01). These findings suggested that mitochondrial autophagy plays a protective role in mitigating NLRP3-mediated cellular injury.

- Impact of NLRP3 silencing combined with mitochondrial autophagy regulation on L-NAME-induced cell damage and oxidative stress in HUVECs. (a) Micrographs of TUNEL staining in HUVECs from various groups (at ×200 magnification, scale bar: 50 µm). (b) Statistical analysis of the number of cells stained by TUNEL. (c) LDH release in HUVECs from various groups. (d) ROS expression in HUVECs from various groups (at ×200 magnification, scale bar: 50 µm). (e) Bar graph statistics of ROS levels. (f) JC-1 staining in HUVECs of each group (at ×100 magnification, scale bar: 100 µm). (g) JC-1 fluorescence quantification graph. n = 3, Data are shown as the means ± SD. (✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001, vs. siNLRP3, NS indicates no significance). NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3, L-NAME: Nω-Nitro-L-arginine methyl ester, 3-MA: 3-Methyladenine, Rapa: Rapamycin, DAPI: 4',6-diamidino-2-phenylindole, TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling, LDH: Lactate dehydrogenase, ROS: Reactive oxygen species, JC-1: 5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide, SD: Standard deviation.
JC-1 staining was performed to measure MMP in HUVECs to further assess the protective role of mitophagy in cellular injury [Figure 5f and g]. Compared with the siNC group, the siNLRP3 group exhibited increased red fluorescence, suggesting that NLRP3 silencing improved mitochondrial function. Following 3-MA treatment, red fluorescence decreased while green fluorescence increased. Meanwhile, Rapa enhanced red fluorescence, indicating that Rapa-induced mitophagy preserves the MMP. These findings indicated that mitophagy plays a pivotal role in NLRP3-mediated pyroptosis, and its activation preserves mitochondrial function while attenuating L-NAME-induced endothelial cell injury.
Mechanistic study of NLRP3 silencing combined with mitophagy regulation in pyroptosis and inflammatory cytokine release
This study examined the impact of NLRP3 silencing combined with the mitochondrial autophagy modulators 3-MA and Rapa on L-NAME-induced pyroptosis and inflammation in HUVECs. Immunofluorescence analysis [Figure 6a and b] demonstrated that NLRP3 silencing (siNLRP3 group) reduced GSDMD fluorescence intensity. Meanwhile, 3-MA enhanced GSDMD fluorescence intensity, and Rapa significantly suppressed it. WB and qRT-PCR analyses further confirmed that NLRP3 silencing (siNLRP3 group) downregulated NLRP3, GSDMD, and caspase-1 and reduced the release of pro-inflammatory cytokines IL-1β and IL-18 [Figure 6c-k]. Furthermore, the modulation of mitochondrial autophagy with 3-MA exacerbated pyroptosis and inflammation, but Rapa significantly mitigated these effects [Figure 6] (P < 0.05 or P < 0.01). These findings underscored the critical role of mitochondrial autophagy in regulating NLRP3-driven pyroptosis and inflammation.
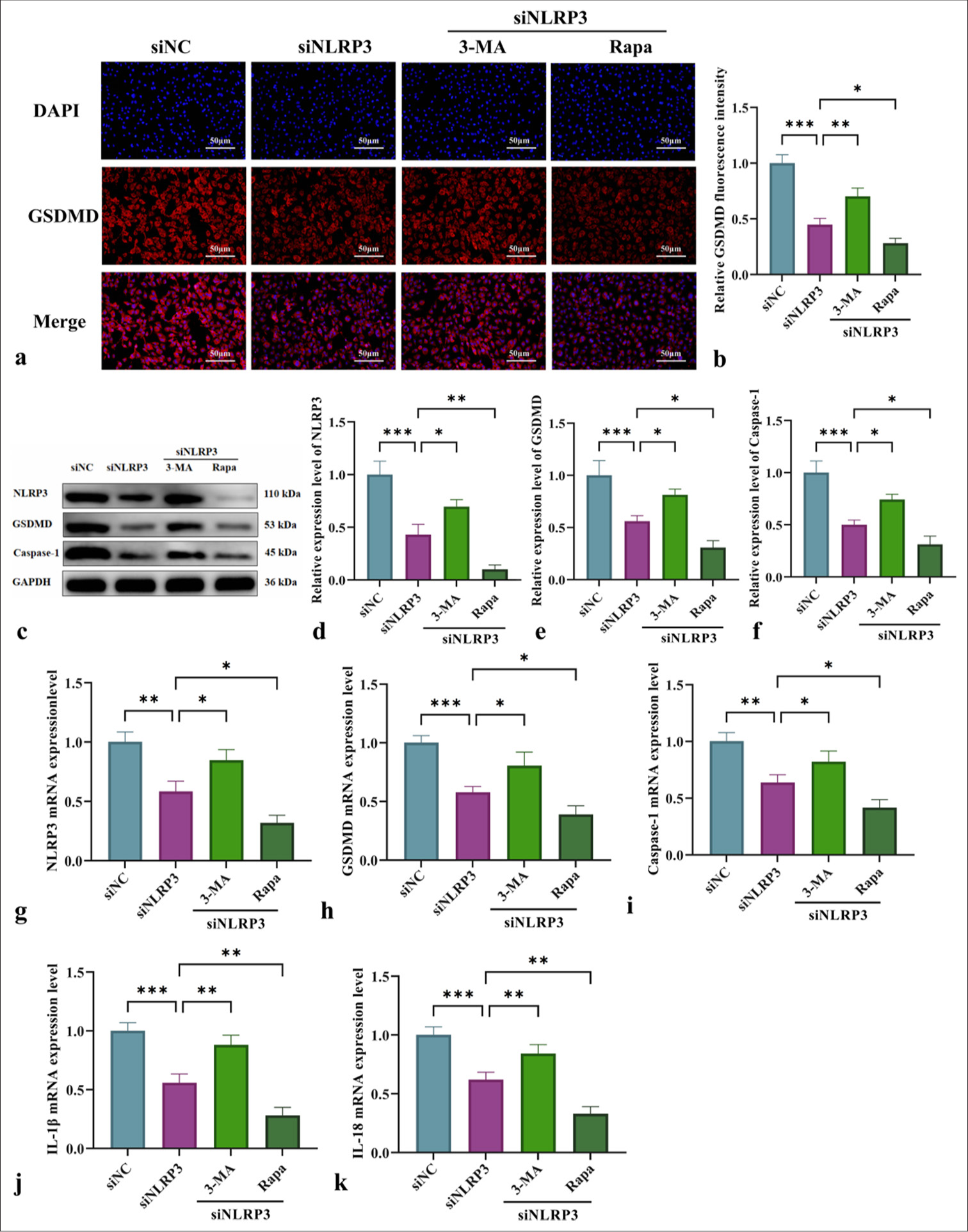
- Exploration of the mechanism of NLRP3 silencing combined with mitochondrial autophagy regulation in pyroptosis and inflammatory cytokine release. (a) Immunofluorescence analysis of GSDMD expression in HUVECs from various groups (at ×200 magnification, scale bar: 50 µm). (b) GSDMD immunofluorescence quantification graph. (c) WB for the detection of NLRP3, GSDMD, and caspase-1 protein levels. (d-f) Quantitative analysis of the content of pyroptosis-related proteins NLRP3, GSDMD, and caspase-1. (g-k) qRT-PCR for the detection of NLRP3, GSDMD, Caspase-1, IL-1β, and IL-18 messenger RNA levels. n = 3, Data are shown as the means ± SD. (✶P < 0.05, ✶✶P < 0.01, ✶✶✶P < 0.001, vs. siNLRP3, NS indicates no significance). 3-MA: 3-Methyladenine, Rapa: Rapamycin, DAPI: 4',6-diamidino-2-phenylindole, NLRP3: Nucleotide-binding oligomerization domain-like receptor protein 3, GSDMD: Gasdermin-D, HUVECs: Human umbilical vein endothelial cells, Caspase-1: Cysteine-aspartic acid protease-1, IL-1β: Interleukin-1β, IL-18: Interleukin-18, WB: Western blot, qRT-PCR: Quantitative real-time polymerase chain reaction, SD: Standard deviation.
DISCUSSION
Hypertensive disorders during pregnancy, including eclampsia, pre-eclampsia, and PIH, are common conditions and contribute significantly to maternal deaths on a global scale.[23] Gestational hypertension is often characterized by proteinuria, edema, and vascular endothelial damage. Endothelial dysfunction is a common and critical complication of various hypertensive disorders, including gestational hypertension.[24] Pregnant women with hypertension often experience a decline in circulating endothelial and endothelial progenitor cell counts,[25] indicating a strong correlation between gestational hypertension and endothelial cell damage/dysfunction. This phenomenon may be associated with multiple pathophysiological mechanisms, particularly NLRP3.
NLRP3 is a crucial protein involved in various essential physiological processes. As a core component of the NLRP3 inflammasome, it plays a role in multiple biological processes.[26,27] Meanwhile, the NLRP3 inflammasome plays a critical role in host defense against pathogens; its dysregulated activation can lead to unchecked infections, thereby contributing to the onset of metabolic disorders, autoimmune diseases, and neurodegenerative conditions.[28,29] GSDMD, a central effector protein in pyroptosis, is expressed in various tissues, including the spleen, colon, kidneys, and intestines. It can be activated through caspase-1, which itself is triggered by the NLRP3 inflammasome or by the cleavage of caspase-4, -5, and -11 in response to intracellular lipopolysaccharide (LPS).[30] Hence, the precise regulation of NLRP3 inflammasome assembly and its downstream signaling cascades is crucial to appropriately initiate immune responses and antimicrobial activities and simultaneously safeguard against undue tissue damage.[31] In the development of gestational hypertension, NLRP3-driven pyroptosis plays a pivotal role in endothelial cell injury.[32,33] The pyroptotic death of endothelial cells results in a compromised endothelial barrier, and the subsequent release of inflammatory mediators deteriorates the vascular structure and function, ultimately leading to the development and advancement of hypertension and its related complications.[34] In this study, a model of gestational hypertension-induced endothelial cell injury was established to investigate the role of NLRP3-mediated pyroptosis. Results demonstrated that in HUVECs from gestational hypertension, NLRP3 was significantly upregulated, along with increases in caspase-1 activation and GSDMD cleavage. These phenomena indicated the induction of NLRP3-mediated pyroptosis. Further experiments demonstrated that mitophagy induction effectively suppressed NLRP3 activation and pyroptosis.
To date, the complex mechanisms underlying NLRP3 inflammasome activation and the specific roles of associated organelles remain poorly elucidated. Among various hypotheses, a prominent perspective suggests that mitochondrial dysfunction is pivotal in triggering NLRP3 inflammasome activation.[35,36] Mitochondria function as the “energy factories” of the cell, responsible for ATP synthesis, and as key regulators of apoptosis and inflammation.[37] Impairments in mitochondrial function, as evidenced by a reduction in MMP or an increase in ROS production, serve as critical triggers for NLRP3 activation. Mitochondrial dysfunction and ROS generation are crucial for initiating cell death signaling pathways, including NLRP3 assembly, which leads to GSDMD oligomerization and pore formation.[38,39] In addition, mitochondria-derived ROS can directly activate NLRP3 and indirectly facilitate its activation by inducing mitochondrial DNA release, further promoting pyroptosis.[40,41] In PIH endothelial cells, NLRP3-mediated pyroptosis was significantly induced, as evidenced by caspase-1 activation and GSDMD formation, ultimately resulting in cell membrane disruption and cellular content release and exacerbating endothelial damage and inflammation.[42,43] This study demonstrated that in HUVECs, excessive NLRP3 activation was correlated with ROS accumulation and mitochondrial dysfunction, suggesting that mitochondrial impairment serves as a major trigger for NLRP3 activation and pyroptosis. Enhancing mitophagy contributes to mitochondrial homeostasis restoration, ROS reduction, and NLRP3 activation and pyroptosis inhibition. qRT-PCR and WB analyses revealed that autophagy activation reduced caspase-1 cleavage products and GSDMD expression, effectively inhibiting the pyroptosis pathway. Meanwhile, mitophagy suppressed the release of inflammatory factors, improved the inflammatory microenvironment, and alleviated endothelial damage in gestational hypertension.
An NLRP3-transfected HUVEC model was established to investigate the functional involvement of NLRP3 in pyroptosis and further elucidate its role in PIH-related endothelial cell damage. The findings indicated that NLRP3 silencing alleviated PIH-related endothelial cell damage and enhanced cell survival by regulating mitochondrial autophagy. The activation of mitochondrial autophagy effectively suppressed NLRP3 inflammasome activation, pyroptosis, and oxidative stress, thereby mitigating cellular damage and reducing the release of inflammatory factors. Overall, mitochondrial autophagy plays a significant protective role in mitigating PIH-related endothelial cell damage by suppressing NLRP3-mediated pyroptosis. This study highlights the pivotal role of NLRP3 in vascular endothelial cell injury associated with gestational hypertension. NLRP3 activation induces excessive mitophagy, leading to mitochondrial dysfunction and endothelial cell pyroptosis. Inhibiting NLRP3 can restore mitochondrial homeostasis, reduce oxidative stress, preserve endothelial function, and mitigate pathological alterations.
This study provides novel insights into the regulation of mitochondrial function in endothelial cells and offers theoretical support for alleviating endothelial damage in gestational hypertension. Future research should focus on establishing a mouse model to further validate the role of NLRP3 and its regulation of mitophagy. However, this study has certain limitations, including its failure to consider the potential influence of signaling pathways such as AKT, AMPK, and NF-κB. Future investigations should further examine signaling pathways such as AKT to achieve a comprehensive understanding of the pathological mechanisms underlying gestational hypertension and to construct an integrated and systematic framework for its pathogenesis.
SUMMARY
This study indicates that increased NLRP3 expression significantly contributes to endothelial cell injury in PIH. Mitochondrial autophagy mitigates this damage by suppressing the NLRP3-mediated pyroptosis. These findings suggest a potential therapeutic approach for PIH by modulating mitochondrial autophagy.
AVAILABILITY OF DATA AND MATERIALS
The data and materials that support the findings of this study are available from the corresponding author upon reasonable request.
ABBREVIATIONS
3-MA: 3-Methyladenine
ANOVA: One-way analysis of variance
ATP: adenosine 5'-triphosphate
Caspase-1: Cysteinyl aspartate-specific proteinase 1
Control: The control group
DAPI: 4’,6-diamidino-2-phenylindole
DCFH-DA: diluted 2,7-Dichlorodihydrofluorescein diacetate
FBS: Fetal bovine serum
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
GSDMD: Gasdermin D
GSDMD-N: N-terminal fragment of GSDMD
HUVEC: Human umbilical vein endothelial cell
IL-18: Interleukin 18
IL-1β: Interleukin 1β
JC-1: 5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcar bocyanine iodide
LC3: Microtubule-associated protein 1 light chain 3
LDH: Lactate dehydrogenase
L-NAME: Nω-Nitro-L-arginine methyl ester
LPS: Lipopolysaccharide
MMP: Mitochondrial membrane potential
MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
NLRP3: Nucleotide-binding oligomerization domain-like
receptor protein 3
NO: Nitric oxide
P62: Sequestosome 1
PBS: Phosphate-buffered saline
PIH: Pregnancy-induced hypertension
pro-IL-18: Pro-interleukin-18
pro-IL-1κ: Pro-interleukin-1κ
qRT-PCR: Quantitative real-time polymerase chain reaction
Rapa: Rapamycin
ROS: Reactive oxygen species
siNLRP3: NLRP3 small interfering RNA
TUNEL: Terminal deoxynucleotidyl transferase dUTP nick
end labeling
WB: Western blot
AUTHOR CONTRIBUTIONS
MSZ: Data curation, investigation, formal analysis, and writing original draft; XZZ: Methodology, formal analysis, writing original draft, writing review, and editing; LLM: Methodology, writing review, and editing. All authors read and approved of the final manuscript. All authors meet ICMJE authorship requirements.
ACKNOWLEDGMENT
We are grateful to the experimental center for its assistance.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval and consent to participate is not required as this study does not involve animal or human experiments.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING: Not applicable.
References
- P360 hypertension during pregnancy. Eur Heart J Suppl. 2022;24:suac012.347.
- [CrossRef] [Google Scholar]
- Sodium, the vascular endothelium, and hypertension: A narrative review of literature. Cardiol Rev 2025 doi: 10.1097/CRD.0000000000000854. [Epub ahead of print]
- [CrossRef] [PubMed] [Google Scholar]
- Abstract 500: Sigmar1 deficiency promotes inflammatory signals by altering nitric oxide bioavailability and mitochondrial redox balance in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2023;43:A500.
- [CrossRef] [Google Scholar]
- Emerging insights into the pathogenesis and therapeutic strategies for vascular endothelial injury-associated diseases: Focus on mitochondrial dysfunction. Angiogenesis. 2024;27:623-39.
- [CrossRef] [PubMed] [Google Scholar]
- NLRP3 licenses NLRP11 for inflammasome activation in human macrophages. Nature Immunol. 2022;23:892-903.
- [CrossRef] [PubMed] [Google Scholar]
- Influenza A virus infection activates NLRP3 inflammasome through trans-Golgi network dispersion. Viruses. 2022;14:88.
- [CrossRef] [PubMed] [Google Scholar]
- NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653-68.
- [CrossRef] [PubMed] [Google Scholar]
- Correction: SARS-CoV-2 S protein activates NLRP3 inflammasome and deregulates coagulation factors in endothelial and immune cells. Cell Commun Signal. 2024;22:64.
- [CrossRef] [Google Scholar]
- iNOS regulates activation of the NLRP3 inflammasome through the sGC/cGMP/PKG/TACE/TNF-α axis in response to cigarette smoke resulting in aortic endothelial pyroptosis and vascular dysfunction. Int Immunopharmacol. 2021;101:108334.
- [CrossRef] [PubMed] [Google Scholar]
- AIP1 regulates ocular angiogenesis via NLRP12-ASC-caspase-8 inflammasome-mediated endothelial pyroptosis. Adv Sci (Weinh). 2024;11:e2405834.
- [CrossRef] [PubMed] [Google Scholar]
- Time-of-day control of mitochondria regulates NLRP3 inflammasome activation in macrophages. FASEB J. 2024;38:e70235.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial DNA in NLRP3 inflammasome activation. Int Immunopharmacol. 2022;108:108719.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondria and NLRP3: To die or inflame. Immunity. 2025;58:5-7.
- [CrossRef] [PubMed] [Google Scholar]
- NLRP3 inflammasome as therapeutic targets in inflammatory diseases. Biomol Ther (Seoul). 2023;31:395-401.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int J Biochem Cell Biol. 2021;136:106013.
- [CrossRef] [PubMed] [Google Scholar]
- Bisphenol A induces pyroptotic cell death via ROS/NLRP3/Caspase-1 pathway in osteocytes MLO-Y4. Food Chem Toxicol. 2022;159:112772.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondria break free: Mitochondria-derived vesicles in aging and associated conditions. Ageing Rese Rev. 2024;102:102549.
- [CrossRef] [PubMed] [Google Scholar]
- The role of autophagy and mitophagy in cancers. Arch Physiol Biochem. 2022;128:281-9.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705.
- [CrossRef] [PubMed] [Google Scholar]
- Recruitment of pro-IL-1α to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc Natl Acad Sci U S A. 2021;118:e2015632118.
- [CrossRef] [PubMed] [Google Scholar]
- 1,8-Cineole ameliorates endothelial injury and hypertension induced by L-NAME through regulation of autophagy via PI3K/mTOR signaling pathway. Eur J Pharmacol. 2023;954:175863.
- [CrossRef] [PubMed] [Google Scholar]
- Global causes of maternal death: A WHO systematic analysis. Lancet Glob Health. 2014;2:e323-33.
- [CrossRef] [PubMed] [Google Scholar]
- Perivascular adipose tissue: A central player in the triad of diabetes, obesity, and cardiovascular health. Cardiovasc Diabetol. 2024;23:455.
- [CrossRef] [PubMed] [Google Scholar]
- Circulating endothelial cells, circulating endothelial progenitor cells, and von Willebrand factor in pregnancies complicated by hypertensive disorders. Am J Reprod Immunol. 2017;77
- [CrossRef] [PubMed] [Google Scholar]
- Hydrogen sulfide plays an important role by influencing NLRP3 inflammasome. Int J Biol Sci. 2020;16:2752-60.
- [CrossRef] [Google Scholar]
- The NLRP3 inflammasome: A therapeutic target for inflammation-associated cancers. Expert Rev Clin Immunol. 2020;16:175-87.
- [CrossRef] [PubMed] [Google Scholar]
- NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18:2114-27.
- [CrossRef] [PubMed] [Google Scholar]
- New insights on NLRP3 inflammasome: Mechanisms of activation, inhibition, and epigenetic regulation. J Neuroimmune Pharmacol. 2024;19:7.
- [CrossRef] [PubMed] [Google Scholar]
- Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180:941-55.e20.
- [CrossRef] [PubMed] [Google Scholar]
- Qiji shujiang granules alleviates dopaminergic neuronal injury of Parkinson's disease by inhibiting NLRP3/caspase-1 pathway mediated pyroptosis. Phytomedicine. 2023;120:155019.
- [CrossRef] [PubMed] [Google Scholar]
- Esculetin inhibits the pyroptosis of microvascular endothelial cells through NF-κB/NLRP3 signaling pathway. Arch Biochem Biophys. 2022;720:109173.
- [CrossRef] [PubMed] [Google Scholar]
- Arctigenin mitigates vascular endothelial injury in rats with pregnancy-induced hypertension via autophagy-NLRP3 inflammasome pathway. Zhongguo Zhong Yao Za zhi. 2023;48:3022-31.
- [Google Scholar]
- Immune and metabolic mechanisms of endothelial dysfunction. Int J Mol Sci. 2024;25:13337.
- [CrossRef] [PubMed] [Google Scholar]
- Role and mechanism of ROS scavengers in alleviating NLRP3-mediated inflammation. Biotechnol Appl Biochem. 2019;66:4-13.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial dysfunction in diabetic neuropathy: Impaired mitophagy triggers NLRP3 inflammasome. Mitochondrion. 2024;79:101972.
- [CrossRef] [PubMed] [Google Scholar]
- Advances in crosstalk among innate immune pathways activated by mitochondrial DNA. Heliyon. 2024;10:e24029.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic regulators of enigmatic inflammasomes in autoimmune diseases and crosstalk with innate immune receptors. Immunology. 2021;163:348-62.
- [CrossRef] [PubMed] [Google Scholar]
- Control of gasdermin D oligomerization and pyroptosis by the ragulator-rag-mTORC1 pathway. Cell. 2021;184:4495-511.e19.
- [CrossRef] [PubMed] [Google Scholar]
- SIRT3 deficiency promotes lung endothelial pyroptosis through impairing mitophagy to activate NLRP3 inflammasome during sepsis-induced acute lung injury. Mol Cell Biol. 2025;45:1-16.
- [CrossRef] [PubMed] [Google Scholar]
- Aluminum activates NLRP3 inflammasome-mediated pyroptosis via reactive oxygen species to induce liver injury in mice. Chem Biol Interact. 2022;368:110229.
- [CrossRef] [PubMed] [Google Scholar]
- Neutrophil extracellular traps induce pyroptosis of pulmonary microvascular endothelial cells by activating the NLRP3 inflammasome. Clin Exp Immunol. 2024;217:89-98.
- [CrossRef] [PubMed] [Google Scholar]
- Purinergic P2X7 receptor mediates hyperoxia-induced injury in pulmonary microvascular endothelial cells via NLRP3-mediated pyroptotic pathway. Open Med (Wars). 2024;19:20241097.
- [CrossRef] [PubMed] [Google Scholar]




