


Translate this page into:
N-acetyltransferase 10 regulates UNC-51-like kinase 1 to reduce tubular cell injury and kidney stone formation

*Corresponding author: Ben Ke, Department of Nephrology, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, China. keben-1989125@163.com
-
Received: ,
Accepted: ,
How to cite this article: Wang L, Huang J, Song L, Ke B. N-acetyltransferase 10 regulates UNC-51-like kinase 1 to reduce tubular cell injury and kidney stone formation. CytoJournal. 2024;21:68. doi: 10.25259/Cytojournal_72_2024
Abstract
Objective:
Among the most common chronic kidney diseases, kidney stones are second only to hypertension. Kidney stones pose a public health threat due to their increased incidence, high recurrence rate, and heavy economic burden. In this work, we investigated the potential mechanism of N-acetyltransferase 10 (NAT10) in oxidative stress and pyroptosis of renal tubular epithelial cells (RTECs).
Material and Methods:
A kidney-stone cell model was simulated using calcium oxalate monohydrate (COM) in vitro. Western blot analysis of NAT10 expression and N4-acetylcytidine RNA immunoprecipitation verified the regulatory efficacy of NAT10 in Unc-51 like autophagy activating kinase 1 (ULK1) ac4C modification. The luciferase reporter gene assay further verified the interaction between NAT10 and ULK1. A kidney stone model was established using BALB/c mice injected with glyoxylic acid.
Results:
COM can dose-dependently suppressed the cell viability and superoxide dismutase activity of HK-2 cells and promoted the release of lactate dehydrogenase and malondialdehyde levels (P < 0.05). COM also promoted apoptosis in HK-2 cells, upregulated the protein levels of caspase-1 and gasdermin D-N, and simultaneously enhanced the HK-2 cell secretion of interleukin-1b (IL-1b) and IL-18 (P < 0.05). The overexpression of NAT10 in HK-2 cells reversed the aforementioned effects, and that of NAT10 upregulated the messenger RNA (mRNA) levels of ULK1 and increased ac4C modification (P < 0.01). Furthermore, only the luciferase activity of the wild-type ULK1 containing NAT10 binding sites was enhanced with the upregulation of NAT10 (P < 0.001). Actinomycin D treatment showed that NAT10 overexpression extended the half-life of ULK1 mRNA (P < 0.01). Silencing of ULK1 neutralized the effects of NAT10 overexpression on COM-induced cell injury (P < 0.05). In addition, the increased expression of NAT10 inhibited crystal deposition, oxidative stress, and apoptosis in vivo (P < 0.05).
Conclusion:
This study confirmed that NAT10 inhibits RTECs oxidative stress and cell pyrodeath through the enhanced ac4C modification of ULK1 and impedes kidney stone progression.
Keywords
N-acetyltransferase 10
N4-acetylcytidine
UNC-51-like kinase 1
Renal tubular epithelial cells
Kidney stones

INTRODUCTION
Nephrolithiasis is a leading disease of the urinary system, and it is caused by the deposition of inorganic salt crystals and organic macromolecules in the kidney parenchyma or pelvic cavity.[1,2] A large number of patients with kidney stone are symptomless in the long term, but a rise in the occurrence of pain, urinary infection, and obstruction has been observed. More than 50% of cases are estimated to relapse after 5 years.[3] The worldwide incidence of kidney stone has soared in the past decades, and it may be attributed to environmental elements, such as an unbalanced diet and limited physical activity.[4] Nephrolithiasis is controlled by multiple factors. Age, gender, ethnicity, and dietary habits influence the prevalence and incidence of this condition. In addition, various risk factors, including obesity, metabolic syndrome, cardiovascular disease, and chronic kidney disorder, are associated with nephrolithiasis.[4-6] Conversely, kidney stones can also act as inducers of these systemic diseases.[7] Given that nephrolithiasis is a global health problem, its pathogenesis must be elucidated, and novel therapeutic strategies must be developed.
Calcium oxalate (CaOx) calculi are the most common type of kidney stone, and they account for approximately 80% of all nephrolithiasis cases.[8] Further, the oxidative stress caused by high oxalate concentrations results in renal tubule injury and calculi formation.[9] Renal tubular epithelial cells (RTECs) injury plays a vital role in kidney stone.[10,11] In terms of mechanism, the key to calculi formation is the adhesion of CaOx crystals to RTECs, which triggers oxidative stress and the overwhelming release of inflammatory factors; this condition results in intrarenal inflammation and RTECs damage.[9,12] Pyroptosis, an inflammatory category of programmed cell death, is usually initiated by NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome and carried out by gasdermin proteins.[13] Accumulating evidence has revealed the importance of cell pyroptosis in a wide range of kidney diseases.[14] Emerging studies have demonstrated the strong association of RTECs pyroptosis with the development of nephrolithiasis.[15,16] Hence, the mechanism governing RTEC pyroptosis must be investigated.
N-Acetyltransferase 10 (NAT10) is the first discovered acetyltransferase involved in N4-acetylcytidine (ac4C) modification, which is a highly conserved RNA modification that broadly regulates cellular activities.[17,18] Although plenty of research has identified NAT10 as a core mediator in a variety of malignant tumors, including pancreatic cancer, bladder carcinoma, colorectal cancer, and gastric carcinoma, its potential in nephrolithiasis remains unknown.[19-21] NAT10 serves as an inhibiting factor for neutrophil pyroptosis in sepsis.[22] Accordingly, this study aimed to probe the participation and mechanism of NAT10 in RTECs pyroptosis to provide new treatment strategies for kidney stones.
MATERIAL AND METHODS
Cell culture
HK-2 cells (CRL-2190), as RTECs, were purchased from the U.S. Typical Culture Preservation Center (ATCC, Manzas, Virginia, United States of America [USA]) and identified by specialized technology resources in Dulbecco’s Modified Eagle Medium (Gibco; 12491015; Invitrogen; USA), supplemented with 10% fetal bovine serum (Gibco; A5670701; Invitrogen; USA), 100 mg/mL streptomycin (Gibco; 15070063; Invitrogen; USA), and 100 U/mL penicillin (Gibco; 15070063; Invitrogen; USA) at 37°C and 5% CO2. The HK-2 cells were found to be negative for mycoplasma contamination.
Cell treatment
For the establishment of a kidney-stone cell model, HK-2 cells were exposed to different doses of CaOx monohydrate (COM) (Yuanye; S66467; China) (0, 0.5, 1, and 2 mM) for 24 h. To upregulate NAT10, we obtained the NAT10-overexpressing vectors and negative control empty plasmids from BersinBio (Guangzhou, China). Cell Counting Kit-8 (CCK-8), lactate dehydrogenase (LDH), and superoxide dismutase (SOD) levels in the cell supernatant were detected to verify model construction.[8] For silencing of Unc-51 like autophagy activating kinase 1 (ULK1), short hairpin RNAs (shRNAs) against ULK1 (termed as shULK1) and scrambled shRNAs (shNC) were designed and produced by GenePharma (Shanghai, China). The shNC sequence is as follows: 5'- CCGGCGTGTCAAGAGAGCATCGTTACTCGAGT AACGATGCTCTCTTGACACGTTTTTG-3'. The shULK1 sequence is as follows: 5'-CCGGACATCGA GAACGTCACCAAGTCTCGAGACTTGGTGACGTTC TCGATGTTTTTT-3'. RTECs were transfected with these plasmids with the assistance of Lipofectamine 2000 (Gibco; 11668500; Invitrogen, USA).
CCK-8
The proliferative capacity of HK-2 cells was estimated using CCK-8 (Beyotime; C0037; China), in accordance with the supplier’s instructions. HK-2 cells were transferred to a 96-well plate and mixed with the CCK-8 reagent (10 mL/well) at 37°C for 2 h. Absorbance was detected at 450 nm using a Tecan Sunrise (Tecan, Switzerland).
Cell apoptosis detection
RTECs were evaluated through flow cytometry using an Annexin V-fluorescein isothiocyanate (FITC) and propyl iodide (PI) double-staining kit (Beyotime; C1062S; China). After various treatments, the RTEC suspension was obtained with trypsin (2–5 min) and incubated with annexin V-FITC for 20 min away from light and retained with PI for an additional 5 min in the dark, as recommended by the manufacturer. Finally, the ratio of apoptotic RTECs was quantified through flow cytometry (FACSCalibur, BD Biosciences, USA).
Enzyme-linked immunosorbent assay (ELISA)
ELISA detection was implemented to examine the expression levels of LDH (Beyotime; P0393S; China), malondialdehyde (MDA) (Beyotime; S0131M; China), SOD (Beyotime; S0101S; China), and glutathione (Beyotime; S0057S; China). The release of interleukin-1b (IL-1b) (R&D Systems; PMLB00C; USA) and IL-18 (R&D Systems; DY318-05; USA) was also assessed by ELISA, following the manufacturer’s protocols.
Western blot
Total protein was extracted from RTECs and kidney samples by using radioimmunoprecipitation assay lysis buffer (Beyotime; P0013B; China). Protein samples (50 mg/well) were loaded onto a 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis gel, transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore; ISEQ00010; USA), and blocked with 5% skim milk at room temperature (RT) for 2 h. Under 4°C, then the membrane against, gasdermin D (GSDMD), caspase-1-N (Cell Signaling Technology, #24232, 1:1000), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology; 8986S; USA) primary antibody mixed overnight at RT during a second antibody detection for 1–2 h. Chemiluminescence detection was performed using an Enhanced Chemiluminescence (ECL) detection kit (Millipore, WBULS0100; USA) with a ChemiDoc MP Imaging System (12003154, Bio-Rad Laboratories, Hercules, CA, USA). GAPDH is an inherent standard for standardization. NAT10 (Abcam, ab194297, 1:1000), ULK1 (Cell Signaling Technology, #8054, 1:1000), caspase-1 (Cell Signaling Technology, #83383, 1:1000), GSDMD-N (Abcam, ab209845, 1:1000), GAPDH (Cell Signaling Technology, #97166, 1:1000), antirabbit secondary antibody (Cell Signaling Technology, #7074, 1:2000), and antimouse secondary antibody (Cell Signaling Technology, #7076, 1:2000) were also used. Western blot analysis was performed using ImageJ (NIH; USA) software and the gray values of the bands were quantified to reflect protein expression.
Real-time fluorescence quantitative polymerase chain reaction (RT-qPCR)
Total RNA isolation was performed using TRIzol solution (Thermo Fisher Scientific; 15596018CN; USA). In compliance with product guidelines, a PrimeScript RT reagent kit (TaKaRa; RR047Q Japan) was adopted for cDNA synthesis. RNA samples were subjected to qPCR analysis in the Applied Biosystems 7500 PCR apparatus (Applied Biosystems, USA) by utilization of a SYBR Premix Ex Taq II kit (Takara; CN830S; Japan). The following specific primers were employed: NAT10, 5’-ATAGCAGCCACAAACATTCGC-3’ (F), 5’-ACACACATGCCGAAGGTATTG-3’ (R); ULK1, 5’-CCCCAACCTTTCGGACTT-3’ (F), 5’-CCAACAGGGTCAGCAAACTC-3’ (R); GAPDH, 5’-AGTGCCAGCCTCGTCTCATA-3’(F), 5’-GGTAACCAGGCGTCCGATAC-3’ (R). In accordance with the 2−ΔΔCq method, relative messenger RNA (mRNA) expressions were determined by normalizing them to the loading control (GAPDH).
ac4C dot blot
After the denaturation of RNA extracts at 75°C, the total RNA samples were cooled with ice, transferred to Hybond-N+ membranes, and subjected to ultraviolet (UV) crosslinking. The membranes were washed under gentle oscillation for 5 min to remove unbound RNAs, followed by blockage in 5% skim milk, probing by primary antibody for ac4C (Abcam, ab252215; Abcam PLC; USA) overnight at 4°C, and treatment with appropriate horse radish peroxidase-labeled secondary antibody for 1.5 h at RT. Ultimately, the blots were visualized using an ECL Detection Kit.
ac4C-RNA immunoprecipitation (ac4C-RIP)
The Magna RIP Kit (Millipore; MAGNARIP03; USA) was used to determine the interaction between NAT10 and ULK1 in the ac4C-RIP experiment. Following the treatment with RIP lysis buffer, the lysates of transfected RTECs were segmented through ultrasonication, and RIP was carried out using anti-ac4C antibody (Abcam, ab253039; Abcam PLC; USA) and negative control IgG (Abcam; 172730; Abcam PLC; USA; 1:2000). After 4 h of incubation with protein A/G magnetic beads (Pierce, USA) at 4°C, bound RNAs were harvested using an elution buffer, purified, and analyzed through qPCR assay.
mRNA stability assay
To determine the potential of NAT10 in ULK1 stability, we conducted qPCR to measure the mRNA expression of ULK1 in NAT10-upregulated HK-2 cells following actinomycin D treatment. The transfected RTECs were obtained and then administered with 200 nM actinomycin D (GLPBIO; GC16866 Cytiva; USA) to suppress RNA synthesis. After 0, 4, 8, and 12 h, the RTECs were subjected to qPCR analysis to detect ULK1 mRNA stability.
Luciferase reporter assay
The 3’ untranslated region (3’UTR) of wild-type (WT) ULK1 was ligated into pGL3 luciferase plasmids (Promega; E175A; Bill Lintion; USA) for ULK1-WT constructs. The binding sites for NAT10 in the ULK1 3’UTR were mutated to generate mutant luciferase vectors (ULK1-MUT). In accordance with the directions for Lipofectamine 2000 (Thermo Fisher; 11668030; USA), the RTECs were cotransfected with the indicated luciferase vectors and NAT10-overexpressing plasmids or negative-control empty plasmids. A Dual-Luciferase Reporter Assay kit (Sanpiatec; 11402JP60; Japan) was used in the detection of luciferase activity as per product protocols.
Animal experiment
The animal research was conducted with the approval of the Laboratory Animal Ethics Committee. Mice (BALB/c, 6–8 weeks old, and 18–22 g) were obtained from GemPharmatech Co, Ltd, China (catalog number 000664) and housed in a specific pathogen-free environment. The temperature was maintained between 20°C–26°C, relative humidity at 40–70%, and a 12 h light/dark cycle. The animals had ad libitum access to sterile, filtered water, and a standard chow diet. A total of 24 mice were used in this study, with 6 mice for each group. To construct a mouse model of kidney stones, we gave the 6-week-old male BALB/c mice an intraperitoneal injection of 80 mg/kg glyoxylic acid (GOX) for 15 days.[8] To verify the effect of NAT10, we inoculated the mice with HK-2 cells infected with either full-length NAT10 lentivirus (LV-NAT10) or GOX-based negative control lentivirus (LV-NC), which were obtained from GENCEFE Biotech (China). All mice were euthanized through rapid cervical dislocation under high CO2 treatment. Kidney tissue was collected and partially immersed in 4% paraformaldehyde for histological analysis and partially frozen using liquid nitrogen for further analysis. The kidney stone model was verified through hematoxylin and eosin (HE) and von Kossa (Abcam, ab150687; Abcam PLC; USA) staining. If COM crystal was observed during histological analysis, the establishment of the model was considered a success.
HE staining
Following fixation, kidney samples were embedded in paraffin and sliced into sections with a thickness of 4 mm. The tissue sections were deparaffinized, hydrated with gradient alcohol, and washed with water. For HE staining, the slices were dyed with hematoxylin (Beyotime; C0105S; China) and rinsed with running water. Then, the sections were treated with 1% hydrochloric acid, counterstained by eosin (Beyotime; C0105S; China), dehydrated, and sealed with neutral gum.
Von Kossa staining
For von Kossa staining, the kidney tissue slices were treated with 1% silver nitrate (Sigma-Aldrich; 209139; USA), exposed to UV radiation for 45 min, rinsed in distilled water, and incubated for 2 min with 5% sodium thiosulphate (MeilunBio, MB2604-1; China). After rinsing with water, the slices were redyed in van Gieson (MeilunBio, PWL228-1; China), dehydrated, and permeabilized with xylene (Sinopharm Chemical Reagent; 1330-20-7; China). Finally, images of HE and von Kossa staining were captured under a light microscope (Zeiss, Germany). ImageJ software was utilized for the quantification of crystal deposition.
Statistical analysis
Data processing was completed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Results were represented as the mean ± standard deviation, and all experiments were performed in three repetitions. Differences among groups were estimated through unpaired Student’s t-test and one-way analysis of variance. Tukey’s Honestly Significant Difference test was conducted as the post hoc test for pairwise comparisons. P < 0.05 was considered statistically significant.
RESULTS
COM led to RTEC damage and a considerable diminution of NAT10 expression
To mimic RTEC injury in vitro, we exposed HK-2 cells to increasing doses of COM (0, 0.5, 1, and 2 mM) for 24 h. CCK-8 assay revealed the decreased viability of HK-2 cells with the increment in COM concentration [Figure 1a]. Compared with the 0 nM group, cell viability showed statistical differences after the treatment with 1 and 2 mM COM (P < 0.01 and P < 0.001, respectively). As displayed in [Figure 1b-d], high doses (1 and 2 mM) of COM enhanced the expressions of LDH and MDA but decreased SOD activity (P all < 0.01). Flow cytometry revealed the gradual elevation in the ratio of apoptotic HK-2 cells with the increase in COM concentration [Figure 1e and f]. Consistently, Western blot validated that COM stimulation contributed to the augmentation of caspase-1 and GSDMD-N expressions in a dose-dependent manner [Figure 1g and h]. Caspase-1 and GSDMD-N showed evident increases compared with that observed at 0 mM (P < 0.05). In addition, the administration of COM promoted the release of IL-1b and IL-18 [Figure 1i and j]. The concentrations of IL-1b and IL-18 were significantly increased after the treatment with 0.5, 1-, and 2-mM COM (P < 0.05). [Figure 1k and l] show that COM weakened the total ac4C modification of HK-2 cells and reduced the NAT10 messenger RNA level (P < 0.05). These consequences suggest that NAT10 may be involved in RTEC injury through the regulation of ac4C modification.
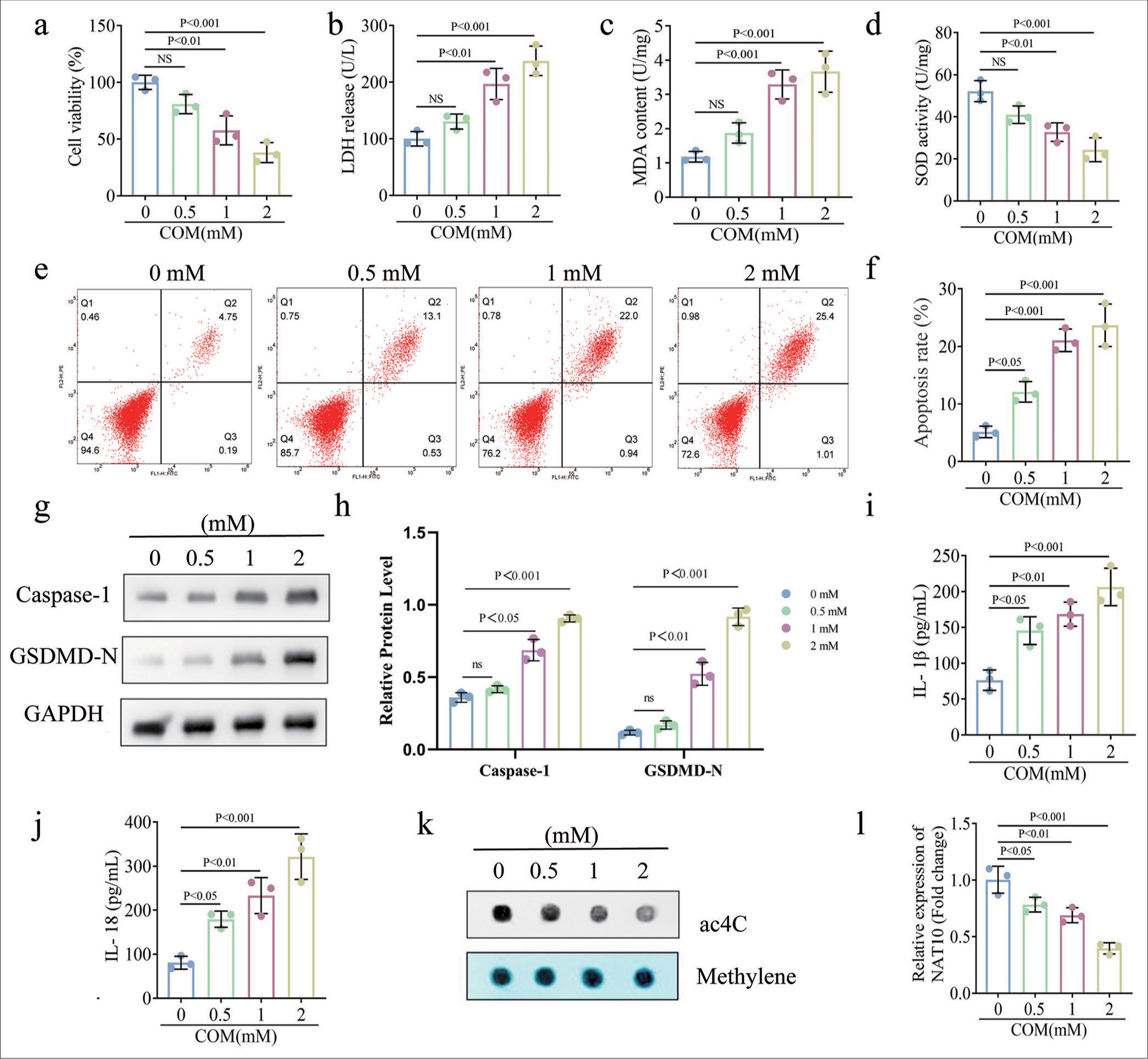
- COM led to RTECs damage and a significant diminution of NAT10 expression. (a) Cell viability (%) of HK-2 after treatment with concentrations of COM (0, 0.5, 1, and 2 mM). (b-d) ELISA kit was used to detect the concentrations of LDH, MDA, and SOD. (e and f) Flow cytometry was performed to determine the apoptosis rate. (g and h) Expressions of apoptosis-related proteins capase-1 and GSDMD-N were measured through Western blot and quantified. (i and j) ELISA was used to detect IL-1β and IL-18 release. (k) ac4C dot blot was used to detect the total ac4C modification level. (l) qPCR analysis of NAT10 expression in kidney-stone cell models. (All the above experiments were repeated 3 times.). COM: Calcium Oxalate Monohydrate; RTECs: Renal tubular epithelial cells; NAT10: N-acetyltransferase 10, ELISA: Enzyme-linked immunosorbent assay, MDA: Malondialdehyde, LDH: Lactate dehydrogenase, SOD: Superoxide dismutase, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, IL: Interleukin, GSDMD-N: Gasdermin D N-terminal, NS: Not significant.
Upregulation of NAT10 alleviates COM-induced oxidative stress and pyroptosis
To further confirm our conclusion, we upregulated NAT10 in the compound-treated RTECs in functional experiments. NAT10 expression was significantly enhanced after transfection [Figure 2a, P < 0.001]. Our observations suggest that the overexpression of NAT10 mitigated the COM-induced decline in HK-2 cell viability [Figure 2b, P < 0.01]. ELISA data elucidate that the ectopic expression of NAT10 led to the restoration of LDH, MDA, and SOD levels in COM-induced HK-2 cells [Figure 2c-e, P < 0.01 for each]. In addition, during NAT10 overexpression, the apoptosis rate of RTECs induced by COM stimulation decreased [Figure 2f and g, P < 0.01 for each]. Similarly, the COM-enhanced expressions of caspase-1 and GSDMD-N were restored due to the up-regulation of NAT10 [Figure 2h and i, P < 0.05 for each]. Consistent with these findings, the overexpression of NAT10 reversed the COM-induced secretion of pro-inflammatory cytokines IL-1b and IL-18 [Figure 2j and k, P < 0.01 for each]. In conclusion, NAT10 inhibited oxidative stress and pyrodeath in COM-induced HK-2 cells.
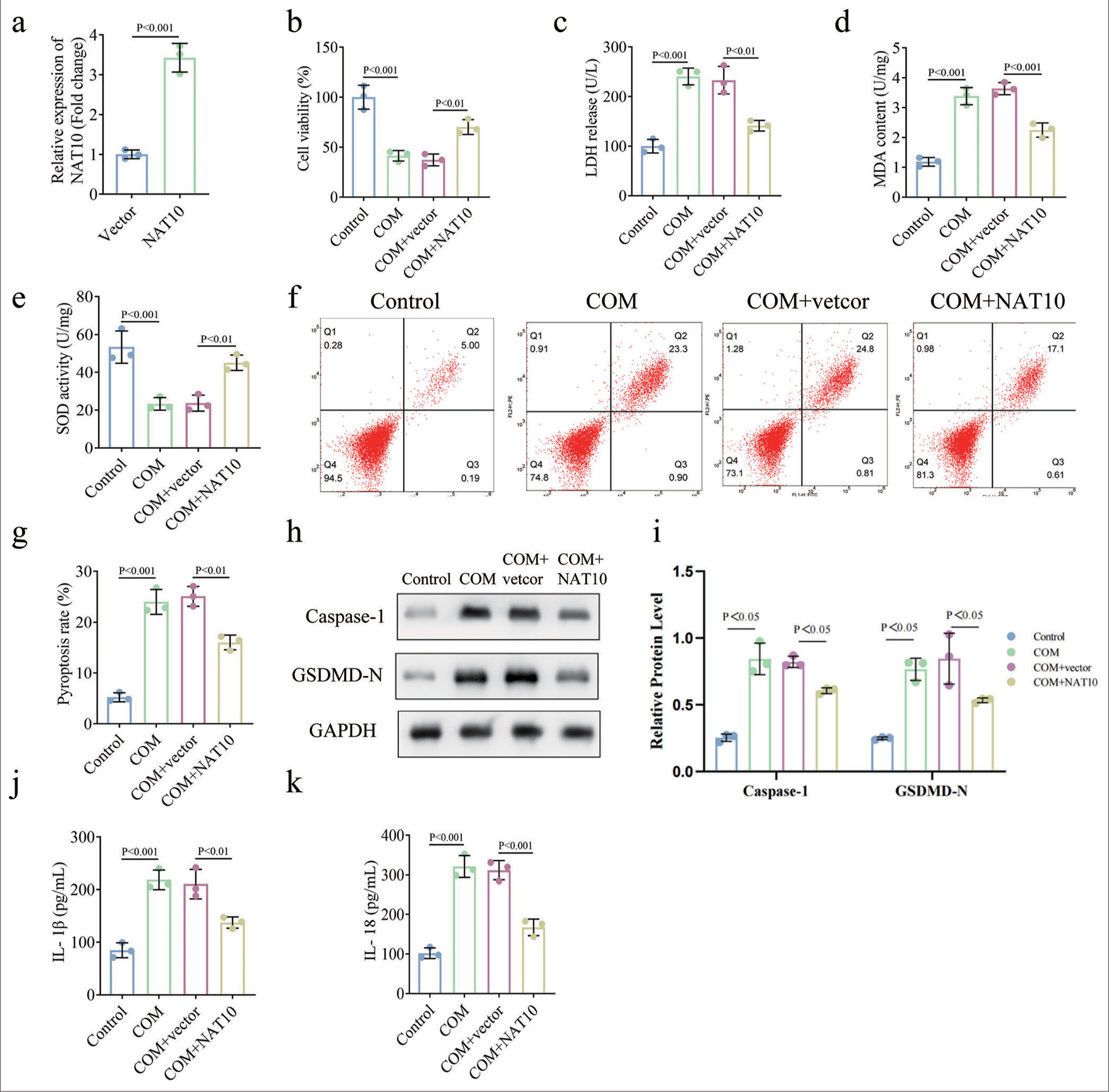
- Upregulation of NAT10 mitigated COM-evoked oxidative stress and apoptosis. To determine the role of NAT10, we administered RETCs with COM and transfected them with a plasmid or empty vector overexpressing NAT10. (a) qPCR was performed to determine transfection efficiency. (b) Cell activity was measured by CCK-8. (c-e) The effect of NAT10 on oxidative stress was evaluated through ELISA. (f and g) Apoptosis rate and the quantification of RTECs were accomplished through flow cytometry. (h and i) Western blot and its quantification were used to detect capase-1 and GSDMD-N levels. (j and k) ELISA was used to detect IL-1β and IL-18 secretion. (All the above experiments were repeated 3 times.). COM: Calcium Oxalate Monohydrate; NAT10: N-acetyltransferase 10, CCK-8: Counting Kit-8, ELISA: Enzyme-linked immunosorbent assay. MDA: Malondialdehyde, LDH: Lactate dehydrogenase, SOD: Superoxide dismutase, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, IL: Interleukin, GSDMD-N: Gasdermin D N-terminal.
NAT10 heightened the expression of ULK1 by inducing its ac4C modification
Next, we sought to determine the relationship of NAT10 with ULK1. qPCR detection proved that NAT10 overexpression prominently heightened the expression of ULK1 [Figure 3a, P < 0.01]. ac4C-RIP assay was completed to certify the role of NAT10 in the ac4c modification of ULK1. Results unravel that NAT10 overexpression enhanced the expression of ac4C-modified ULK1 compared to the control group [Figure 3b, P < 0.01]. Moreover, the luciferase reporter assay confirmed that only the luciferase activity of the WT ULK1 harboring the NAT10 binding sites was strengthened in response to NAT10 upregulation [Figure 3c, P < 0.001]. Afterward, we investigated the effects of NAT10 on ULK1 mRNA stability using actinomycin D. qPCR analysis illustrated that the ectopic expression of NAT10 extended the half-life of ULK1 under actinomycin D treatment [Figure 3d, P < 0.01]. Thus, NAT10 facilitated ac4C modification of ULK1 to increase its mRNA stability.
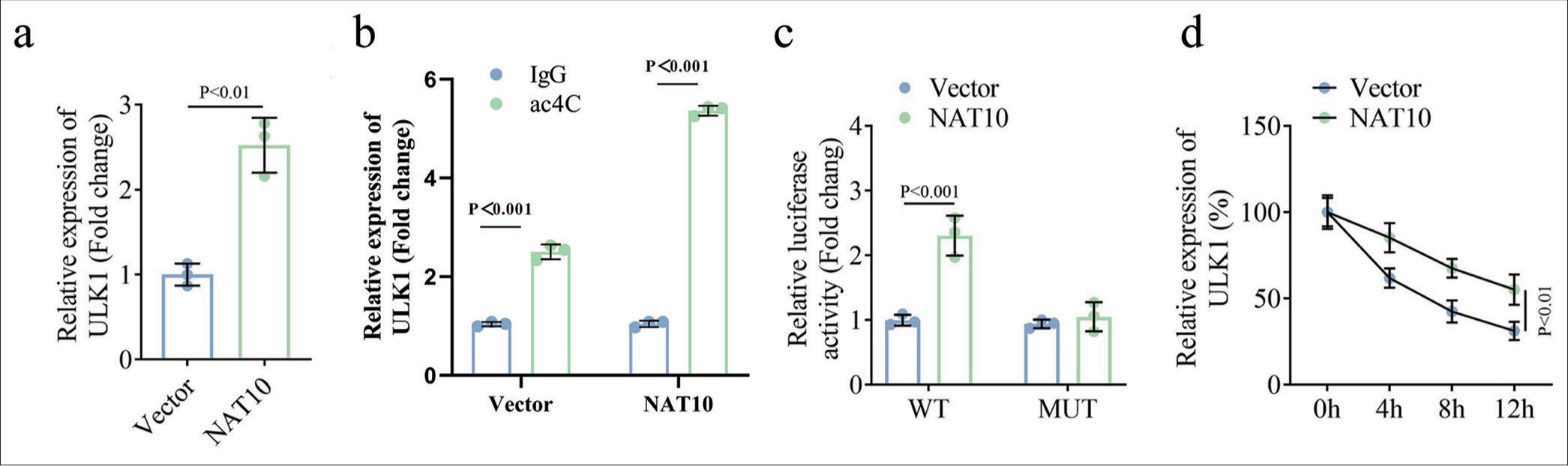
- NAT10 heightened the expression of ULK1 by inducing its ac4C modification. (a) qPCR assay was conducted to identify the expression of ULK1 in response to NAT10 overexpression. (b) ac4C-RIP was conducted to verify the regulatory potency of NAT10 in ULK1 ac4C modification. (c) The interplay of NAT10 with ULK1 was further validated through luciferase reporter assay. (d) The RNA decay assay was applied to inquire the function of NAT10 in mRNA stability of ULK1. (All the above experiments were repeated 3 times.). ULK1: Unc-51 like autophagy activating kinase 1, NAT10: N-acetyltransferase 10, ac4C: N4-acetylcytidine, ac4C-RIP: ac4C-RNA immunoprecipitation, qPCR: Quantitative polymerase chain reaction, IgG: Immunoglobulin G, WT: Wild type, MUT: Mutant.
Knockdown of ULK1 counteracted the promoting effects of NAT10 overexpression on COM-stimulated RTEC injury
Given the indicated information, we silenced ULK1 in NAT10-upregulated RTECs induced by COM. qPCR assay confirmed the transfection efficiency of ULK1 [Figure 4a, P < 0.001]. CCK-8 and ELISA results reveal that knockdown of ULK1 neutralized the ameliorative function of NAT10 overexpression in cell viability and oxidative stress in COM-treated HK-2 cells [Figure 4b-e, P < 0.05 for each]. Flow cytometry analysis showed that the apoptosis rate of COM-induced RTECs decreased by NAT10 upregulation was elevated due to the depletion of ULK1 [Figure 4f and g, P < 0.01 for each]. Coincidently, our data unveil that silencing ULK1 abrogated the influences of NAT10 overexpression on the expressions of pyroptosis-related proteins and cytokines [Figure 4h-j, P < 0.05 for each]. Overall, these findings prove that NAT10 executed a protective role in RTEC injury caused by COM through the enhancement of ULK1 expression.
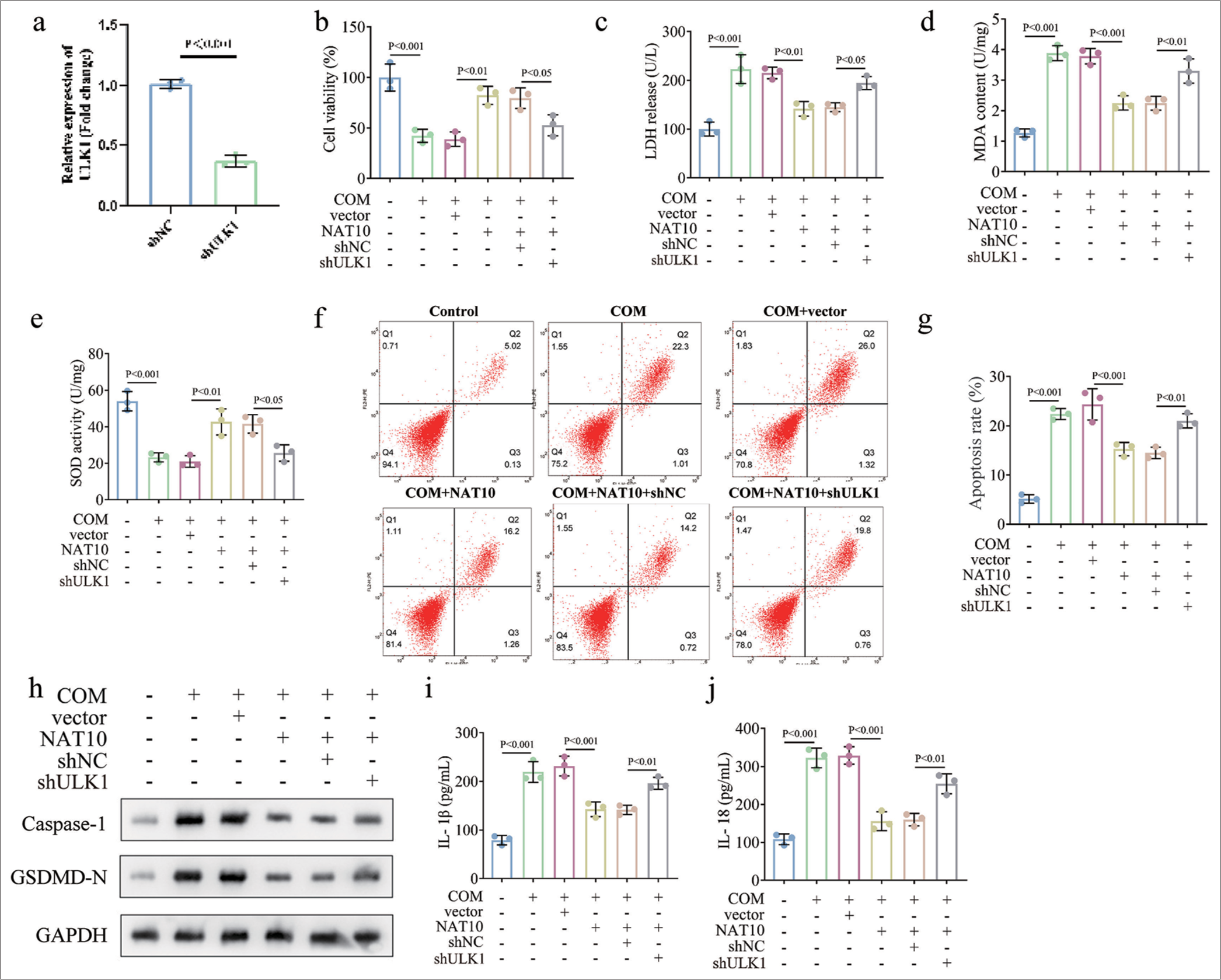
- Knockdown of ULK1 counteracted the promoting effects of NAT10 overexpression on COM-stimulated RTECs injury. To verify the involvement of NAT10/ULK1 axis in CaOx stone formation, we knocked down ULK1 in COM-treated after the upregulation of NAT10. (a) Transfection efficiency certified by qPCR. (b) CCK-8 assay was utilized to estimate cell viability. (c-e) ELISA detection data of LDH, MDA, and SOD expressions. (f and g) Flow cytometry analysis of cell pyroptosis in various groups. (h) Western blot was performed to detect capase-1 and GSDMD-N levels. (i and j) Release of IL-1β and IL-18 as determined by ELISA. (All the above experiments were repeated 3 times.). ULK1: Unc-51 like autophagy activating kinase 1, NAT10: N-acetyltransferase 10, COM: Calcium oxalate monohydrate, RTECs: Renal tubular epithelial cells, CCK-8: Counting Kit-8, LDH: Lactate dehydrogenase, SOD: Superoxide dismutase, ELISA: Enzyme-linked immunosorbent assay, MDA: Malondialdehyde, CaOx: Calcium oxalate, IL: Interleukin, qPCR: Quantitative polymerase chain reaction, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, sh: Short hairpin, GSDMD-N: Gasdermin D N-terminal, sh-NC: sh-negative control.
Overexpression of NAT10 alleviated renal calcinosis in mice
Finally, we constructed a mouse kidney-stone model using GOX. HE and von Kossa staining analyses showed that GOX led to CaOx crystal deposition in mouse kidney tissue, and NAT10 overexpression improved GOX-induced crystal deposition [Figure 5a and b, P < 0.01 for each]. ELISA results reveal that GOX increased the MDA levels and weakened SOD and glutathione (GSH) concentrations in the kidney tissue. Meanwhile, the upregulated NAT10 alleviated the effects of GOX on oxidative stress [Figure 5c-e, P < 0.01 for each]. In addition, Western blotting indicated that GOX enhanced the expressions of caspase 1 and GSDMD-N, which were then restored by the ectopic expression of NAT10 [Figure 5f and g, P < 0.05]. Overall, we demonstrated the obstructive role of NAT10 in the progression of kidney stones in vivo.
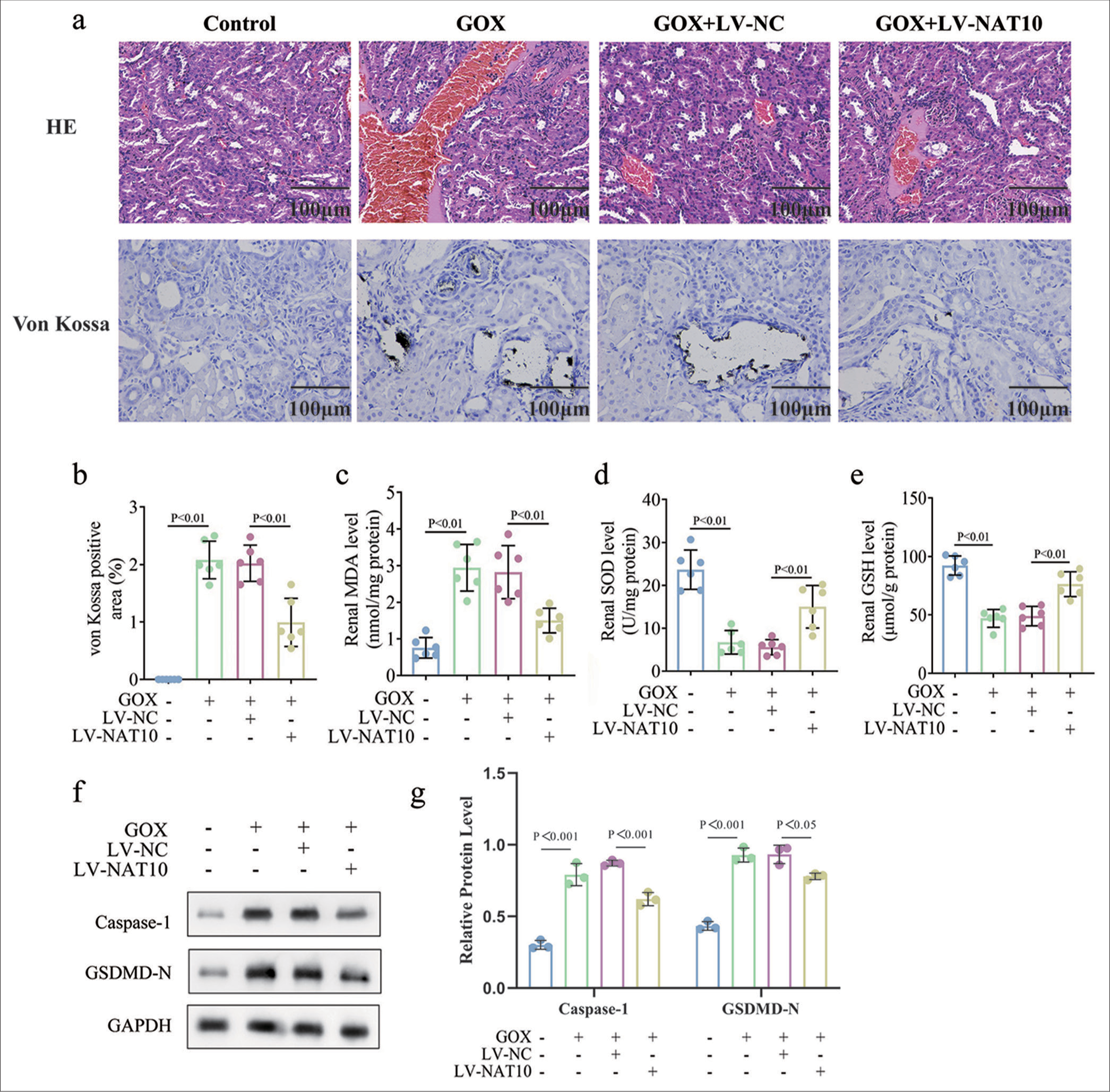
- Enforced expression of NAT10 alleviated nephrocalcinosis in mice. The animal model of kidney stone was established by injecting GOX into BALB/c mice. NAT10 was overexpressed in mouse models to further confirm its function in vivo (n=6). (a) HE and von Kossa staining of kidney tissues in various groups. (b) Quantification of von Kossa positive (crystal deposition) staining in renal samples from different groups. (c-e) The expressions of MDA, SOD, and GSH were examined by ELISA. (f and g) Capase-1 and GSDMD-N levels in renal tissues were measured through Western blot and its quantification. (All the above experiments were repeated 3 times.). NAT10: N-acetyltransferase 10, SOD: Superoxide dismutase, ELISA: Enzyme-linked immunosorbent assay, MDA: Malondialdehyde.
DISCUSSION
Nephrolithiasis is a painful disease with prevalence rates of 8–10% across the world.[12] This condition can progress to serious kidney diseases, such as hydronephrosis, renal insufficiency, chronic kidney disease, and end-stage renal disease.[23,24] Its incidence is incremental in both genders mainly due to changes in the environmental factors caused by social development.[25,26] Despite the discovery of several effective treatments for nephrolithiasis, its high recurrence following the removal of calculi is still a conundrum.[27] In addition, kidney stone treatment is expensive, which generates a huge financial burden on patients’ families and healthcare systems.[28] Thus, the prevention strategy of kidney stone formation must be explored. Given that CaOx supersaturation is a major cause of nephrolithiasis, in this research, RTECs were subjected to the frequently used COM treatment to establish an in vitro model of kidney stone by referring to reported studies.[29-31]
Inflammation is one of the clinical traits of nephrolithiasis, and such a condition indicates a close relationship between the kidney and the immune system.[32] CaOx crystals lead to the activation of NLRP3 inflammasome, which serves as a core sensor for pyroptosis in nephrolithiasis.[33,34] The NLRP3 inflammasome can convert pre-caspase-1 into activated caspase-1 through cleavage, which results in the maturation and secretion of pro-inflammatory IL-1b and IL-18 and RTEC damage and expedites nephrolithiasis progression.[35,36] Oxidative stress is also a critical inducible factor in CaOx crystal-evoked renal injury, which is manifested as the augmentation of LDH and MDA expression and the diminution of antioxidant SOD and GSH levels.[37,38] Herein, our results signify that COM stimulation weakened cell viability, aggravated oxidative stress, and increased cell pyroptosis in RTECs, all of which were in agreement with the findings of previous investigations.[16,39,40]
ac4C is a conserved chemical RNA modification, and it performs a crucial role in the stability of tRNA tertiary structure, rRNA biogenesis, and the improvement of mRNA stability and translation efficiency.[41,42] NAT10 is a well-known “writer” that promotes ac4C production and is involved in modulating the occurrence and development of human diseases.[43,44] We discovered the inhibited ac4C modification was and reduced NAT10 level in COM-treated HK-2 cells. Further experiments confirmed that NAT10 was conducive to the alleviation of COM-triggered RTEC injury.
ULK1 is a kinase responsible for mediating autophagy initiation and functions as a negative regulator of NLRP3 inflammasome.[45,46] Moreover, accumulating evidence has expounded the suppressive role of ULK1 in cell pyroptosis.[47,48] NAT10 targets ULK1 through its mRNA ac4C modification.[22] Consistently, we verified the promoting property of NAT10 in ULK1 expression, which was accomplished by facilitating NAT10-mediated ac4C modification of ULK1. In addition, our study demonstrated that silencing ULK1 abated the protection of NAT10 upregulation in COM-stimulated RTECs. Finally, animal assay ulteriorly justified that NAT10 retarded kidney stone progression in mice through the mitigation of COM deposition, oxidative stress, and pyroptosis.
This study revealed that NAT10 effectively inhibited COM-induced oxidative stress and cell pyrosis in RTECs by enhancing ac4C modification of ULK1, which prevented the formation of kidney stones. The results show that NAT10 overexpression can substantially reverse COM-induced RTEC cell damage, and the silencing of ULK1 eliminated the protective effect of NAT10 overexpression, which confirms the key role of ULK1 in this process. In addition, the enhanced expression of NAT10 in animal models inhibited crystal deposition, oxidative stress, and apoptosis in vivo, which further proves the potential therapeutic effect of NAT10 on kidney stone formation. These findings provide a scientific basis for the development of novel therapeutic strategies and exhibit the potential of NAT10 and its mediated ULK1 ac4C modification as therapeutic targets.
SUMMARY
The present study clarified the essential function of NAT10 in alleviating oxidative stress and pyroptosis in RTECs, which are central to the pathogenesis of kidney stone formation. We have demonstrated that NAT10 exerts its effects by promoting the ac4C modification of ULK1, thereby inhibiting the advancement of kidney stone disease. This conclusion encapsulates our findings and underscores their significance in understanding and potentially managing kidney stone pathophysiology.
AVAILABILITY OF DATA AND MATERIALS
Data to support the findings of this study are available on reasonable request from the corresponding author.
ABBREVIATIONS
ac4C: N4-acetylcytidine
CaOx: Calcium oxalate
CCK-8: Cell Counting Kit-8
COM: Calcium oxalate monohydrate
ELISA: Enzyme-linked immunosorbent assay
FITC: Fluorescein isothiocyanate
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
GSDMD-N: Gasdermin D N-terminal
GSH: Glutathione
HE: Hematoxylin and eosin
HDAC2: Histone deacetylase 2
IgG: Immunoglobulin G
IL: Interleukin
LDH: Lactate dehydrogenase
MDA: Malondialdehyde
MUT: Mutant
NAT10: N-acetyltransferase 10
NLRP3: NOD-like receptor family pyrin domain-containing 3
NS: Not significant
PI: Propidium iodide
PVDF: Polyvinylidene fluoride
qPCR: Quantitative polymerase chain reaction
RIP: RNA immunoprecipitation
RTECs: Renal tubular epithelial cells
sh-NC: Short hairpin RNA negative control
sh-RNA: Short hairpin RNA
SOD: Superoxide dismutase
ULK1: Unc-51-like autophagy activating kinase 1
UV: Ultraviolet
WT: Wild type
AUTHOR CONTRIBUTIONS
LW and BK: Designed the research study; JJH: Performed the research; LS: Collected and analyzed the data. All authors have been involved in drafting the manuscript and all authors have been involved in revising it critically for important intellectual content. All authors give final approval of the version to be published. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The research/study approved by the Institutional Review Board at The Second Affiliated Hospital of Nanchang University, number Approval No.: [2024]NO. (101), dated 2024. The study did not involve human subjects, and patient informed consent is not required.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This research was funded by the Youth Project of Jiangxi Education Department (No.GJJ180149).
References
- Trends in the prevalence of kidney stones in the United States from 2007 to 2016. Urolithiasis. 2021;49:27-39.
- [CrossRef] [PubMed] [Google Scholar]
- Nephrolithiasis In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, eds. Endotext. South Dartmouth, MA: MDText.com, Inc; 2000.
- [Google Scholar]
- Effectiveness of treatment modalities on kidney stone recurrence. Clin J Am Soc Nephrol. 2017;12:1699-708.
- [CrossRef] [PubMed] [Google Scholar]
- Kidney stones and the risk for chronic kidney disease. Clin J Am Soc Nephrol. 2009;4:804-11.
- [CrossRef] [PubMed] [Google Scholar]
- Risk factors for kidney stone disease recurrence: A comprehensive meta-analysis. BMC Urol. 2022;22:62.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of kidney stones. Healthcare (Basel). 2023;11:424.
- [CrossRef] [PubMed] [Google Scholar]
- The role of cell-crystal reaction mediated inflammation in the formation of intrarenal calcium oxalate crystals. Zhonghua Wai Ke Za Zhi. 2018;56:733-6.
- [Google Scholar]
- Erythrocyte oxidative stress in patients with calcium oxalate stones correlates with stone size and renal tubular damage. Urology. 2014;83:510.e9-7.
- [CrossRef] [PubMed] [Google Scholar]
- Overexpression of Mir-30c-5p reduces cellular cytotoxicity and inhibits the formation of kidney stones through Atg5. Int J Mol Med. 2020;45:375-84.
- [CrossRef] [Google Scholar]
- Calcium oxalate crystal adherence to hyaluronan-, osteopontin-, and Cd44-expressing injured/regenerating tubular epithelial cells in rat kidneys. J Am Soc Nephrol. 2003;14:3155-66.
- [CrossRef] [PubMed] [Google Scholar]
- Kidney stone matrix proteins ameliorate calcium oxalate monohydrate induced apoptotic injury to renal epithelial cells. Life Sci. 2016;164:23-30.
- [CrossRef] [PubMed] [Google Scholar]
- Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12:4310-29.
- [CrossRef] [PubMed] [Google Scholar]
- Pyroptosis: A new frontier in kidney diseases. Oxid Med Cell Longev. 2021;2021:6686617.
- [CrossRef] [PubMed] [Google Scholar]
- Long noncoding Rna Linc00339 promotes renal tubular epithelial pyroptosis by regulating the Mir-22-3p/Nlrp3 axis in calcium oxalate-induced kidney stone. J Cell Biochem. 2019;120:10452-62.
- [CrossRef] [PubMed] [Google Scholar]
- Melatonin alleviates intrarenal caox crystals deposition through inhibiting LPS-induced non-canonical inflammasome-mediated renal tubular epithelial cells pyroptosis. Int Immunopharmacol. 2023;124:110796.
- [CrossRef] [PubMed] [Google Scholar]
- Ac4c acetylation of Runx2 catalyzed by Nat10 Spurs osteogenesis of bmscs and prevents ovariectomy-induced bone loss. Mol Ther Nucleic Acids. 2021;26:135-47.
- [CrossRef] [PubMed] [Google Scholar]
- Nat10 promotes cell proliferation by acetylating Cep170 Mrna to enhance translation efficiency in multiple myeloma. Acta Pharm Sin B. 2022;12:3313-25.
- [CrossRef] [PubMed] [Google Scholar]
- NAT10-mediated mRNA N4-acetylcytidine modification promotes bladder cancer progression. Clin Transl Med. 2022;12:e738.
- [CrossRef] [PubMed] [Google Scholar]
- NAT10 promotes liver lipogenesis in mouse through N4-acetylcytidine modification of Srebf1 and Scap mRNA. Lipids Health Dis. 2024;23:368.
- [CrossRef] [PubMed] [Google Scholar]
- N-acetyltransferase 10 promotes colon cancer progression by inhibiting ferroptosis through N4-acetylation and stabilization of ferroptosis suppressor protein 1 (FSP1) mRNA. Cancer Commun (Lond). 2022;42:1347-66.
- [CrossRef] [PubMed] [Google Scholar]
- Nat10 regulates neutrophil pyroptosis in sepsis via acetylating Ulk1 Rna and activating sting pathway. Commun Biol. 2022;5:916.
- [CrossRef] [PubMed] [Google Scholar]
- Kidney stones and kidney function loss: A cohort study. BMJ. 2012;345:e5287.
- [CrossRef] [PubMed] [Google Scholar]
- Nephrolithiasis and loss of kidney function. Curr Opin Nephrol Hypertens. 2013;22:390-6.
- [CrossRef] [PubMed] [Google Scholar]
- Risk of kidney stones: Influence of dietary factors, dietary patterns, and vegetarian-vegan diets. Nutrients. 2020;12:779.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology and economics of nephrolithiasis. Investig Clin Urol. 2017;58:299-306.
- [CrossRef] [PubMed] [Google Scholar]
- Theaflavin protects against oxalate calcium-induced kidney oxidative stress injury via upregulation of Sirt1. Int J Biol Sci. 2021;17:1050-60.
- [CrossRef] [PubMed] [Google Scholar]
- Ferrostatin1 alleviates oxalateinduced renal tubular epithelial cell injury, fibrosis and calcium oxalate stone formation by inhibiting ferroptosis. Mol Med Rep. 2022;26:256.
- [CrossRef] [PubMed] [Google Scholar]
- Immunotherapy for stone disease. Curr Opin Urol. 2020;30:183-9.
- [CrossRef] [PubMed] [Google Scholar]
- Proposal for pathogenesis-based treatment options to reduce calcium oxalate stone recurrence. Asian J Urol. 2023;10:246-57.
- [CrossRef] [PubMed] [Google Scholar]
- An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatr Nephrol. 2016;31:535-44.
- [CrossRef] [PubMed] [Google Scholar]
- The mechanism of Nlrp3 inflammasome activation and its pharmacological inhibitors. Front Immunol. 2022;13:1109938.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammation and kidney stones: Cause and effect? Curr Opin Urol. 2023;33:129-35.
- [CrossRef] [PubMed] [Google Scholar]
- Polydatin protects against calcium oxalate crystal-induced renal injury through the cytoplasmic/mitochondrial reactive oxygen species-Nlrp3 inflammasome pathway. Biomed Pharmacother. 2023;167:115621.
- [CrossRef] [PubMed] [Google Scholar]
- Vitexin exerts protective effects against calcium oxalate crystal-induced kidney pyroptosis in vivo and in vitro. Phytomedicine. 2021;86:153562.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanism of Mirna-141-3p in calcium oxalate-induced renal tubular epithelial cell injury Via Nlrp3-mediated pyroptosis. Kidney Blood Press Res. 2022;47:300-8.
- [CrossRef] [PubMed] [Google Scholar]
- Butyric acid inhibits oxidative stress and inflammation injury in calcium oxalate nephrolithiasis by targeting Cyp2c9. Food Chem Toxicol. 2023;178:113925.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of Nat10 as Ac4c writer in diseases. Mol Ther Nucleic Acids. 2023;32:359-68.
- [CrossRef] [PubMed] [Google Scholar]
- N4-acetylcytidine regulates the replication and pathogenicity of enterovirus 71. Nucleic Acids Res. 2022;50:9339-54.
- [CrossRef] [PubMed] [Google Scholar]
- Acetylation of cytidine in MRNA promotes translation efficiency. Cell. 2018;175:1872-86.e24.
- [CrossRef] [PubMed] [Google Scholar]
- Emerging roles of Rna Ac4c modification and Nat10 in mammalian development and human diseases. Pharmacol Ther. 2023;253:108576.
- [CrossRef] [PubMed] [Google Scholar]
- Ampk/Ulk1-mediated phosphorylation of parkin act domain mediates an early step in mitophagy. Sci Adv. 2021;7:eabg4544.
- [CrossRef] [PubMed] [Google Scholar]
- Sesn2/Sestrin2 suppresses sepsis by inducing mitophagy and inhibiting Nlrp3 activation in macrophages. Autophagy. 2016;12:1272-91.
- [CrossRef] [PubMed] [Google Scholar]
- Short-chain fatty acids attenuate renal fibrosis and enhance autophagy of renal tubular cells in diabetic mice through the HDAC2/ULK1 axis. Endocrinol Metab (Seoul). 2022;37:432-43.
- [CrossRef] [PubMed] [Google Scholar]
- Traf3 promotes ros production and pyroptosis by targeting Ulk1 ubiquitination in macrophages. FASEB J. 2020;34:7144-59.
- [CrossRef] [PubMed] [Google Scholar]




