


Translate this page into:
PR/SET domain 1 targeting glutathione peroxidase 4 regulates chronic hepatitis B liver fibrosis through ferroptosis

Haibing Gao

Huaxi Ma
*Corresponding author: Haibing Gao, Department of Infectious Diseases, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, China. drgaohb605@163.com
Huaxi Ma, Department of Infectious Diseases, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, China. smileforever137@126.com
-
Received: ,
Accepted: ,
How to cite this article: Wu W, Ke W, Shi W, Lin T, Lin S, Lin M, et al. PR/SET domain 1 targeting glutathione peroxidase 4 regulates chronic hepatitis B liver fibrosis through ferroptosis. CytoJournal. 2024;21:78. doi: 10.25259/Cytojournal_123_2024
Abstract
Objective:
Addressing the inhibition and reversal of chronic hepatitis B fibrosis is an urgent global challenge, which highlights the critical need to understand its underlying mechanisms. Inhibiting the activation of hepatic stellate cells (HSCs) is an important strategy for fibrosis reversal. In particular, the induction of ferroptosis in HSCs presents a promising avenue for curtailing liver fibrosis. Therefore, this study explores the influence of PR/SET domain 1 (PRDM1), which is a transcriptional regulator, on the progression of liver fibrosis by regulating HSC ferroptosis through glutathione peroxidase 4 (GPX4).
Material and Methods:
We used protein–protein interaction databases to analyze the interacting proteins of GPX4. The messenger ribonucleic acid levels of PRDM1 and GPX4 in liver tissues with varying degrees of fibrosis were examined using quantitative polymerase chain reaction. Cell lines with interference and overexpression of PRDM1/GPX4 were established. Reactive oxygen species (ROS) activity, malondialdehyde (MDA) concentration, cell proliferation capacity, as well as the expression levels of GPX4, a-smooth muscle actin, vimentin, and desmin, were assessed to investigate the relationship between PRDM1 and hepatic fibrosis, as well as its impact on ferroptosis in HSCs.
Results:
A significant negative correlation was observed between the transcriptional regulator PRDM1 and GPX4. As the degree of fibrosis worsened, PRDM1 decreased significantly, whereas GPX4 increased significantly. The overexpression of PRDM1 markedly increased ROS and MDA concentrations, but it decreased cell proliferation capacity, GPX4 expression levels, and activation marker protein levels. Interference with PRDM1 yielded opposite results. The expression level of GPX4 did not affect PRMD1 expression levels. Compared with cells with single interference of PRDM1, simultaneous interference with PRDM1 and GPX4 significantly inhibited the activity and proliferation capacity of HSCs. It also elevated ROS activity and MDA concentrations. When ferroptosis inhibitors were added, ROS activity and MDA concentrations decreased, and the proliferation capacity and activity of HSCs increased. Opposite results were obtained when PRDM1 and GPX4 were overexpressed simultaneously.
Conclusion:
PRDM1 is implicated in the occurrence and progression of hepatic fibrosis. It may act as an upstream regulatory factor of GPX4, which exerts control over ferroptosis by suppressing the transcription of GPX4. Ultimately, the activation of HSCs is promoted.
Keywords
Ferroptosis
Chronic hepatitis B
Glutathione peroxidase 4
PR/SET domain 1

INTRODUCTION
Chronic hepatitis B (CHB) liver fibrosis results from immune-mediated damage to hepatic tissue caused by persistent hepatitis B virus (HBV) infection, as well as the continued accumulation of extracellular matrix during the repair process.[1] Hepatic fibrosis is a dynamic pathological process and an important consequence of chronic inflammatory liver disease, with the potential to advance to cirrhosis, end-stage liver disease, and hepatocellular carcinoma (HCC) if left untreated.[2] Hepatic fibrosis is believed to be a reversible pathophysiological process in chronic liver disease.[3] Therefore, investigating its reversal mechanisms is crucial.
Hepatic stellate cells (HSCs) are considered the central driving factor of hepatic fibrosis. HSCs can differentiate into myofibroblasts when activated. These myofibroblasts secrete matrix metalloproteinases and other matrix component proteins; this phenomenon initiates tissue remodeling within the liver and triggers various liver cell types to secrete chemokines and inflammatory cytokines, which exacerbate the development of hepatic fibrosis.[4-7] Regression of hepatic fibrosis is accompanied by a reduction or absence of activated HSCs. Therefore, strategies focused on inactivating HSCs are essential for inhibiting the progression of fibrosis.
Ferroptosis is a novel form of iron-dependent regulated cell death, and it differs from apoptosis, necrosis, and autophagy. It occurs when the repair system for glutathione (GSH)-dependent lipid peroxides is compromised, which leads to the lethal accumulation of lipid-based reactive oxygen species (ROS) within cells.[8] This phenomenon leads to the depletion of GSH and reduced activity of glutathione peroxidase 4 (GPX4).[9] Several studies have reported that the progression of liver fibrosis is impacted by ferroptosis. Ferroptosis can increase acetaminophen-induced liver fibrosis in mice, which can be prevented by the presence of ferritin-1.[10] In addition, fibroblast growth factor 21 has been found to inhibit ferroptosis, which mitigates iron overload-induced liver injury and fibrosis.[11] Notably, GPX4, which is a key protein involved in regulating ferroptosis, has a relatively significant function in this pathway. Lin et al. demonstrated that 17a-hydroxypregnenolone acts as an agonist of G protein-coupled receptor 56 to antagonize ferroptosis and efficiently attenuates liver injury before or after insult.[12] In another study, exosome-derived beclin 1 secreted by mesenchymal stem cells was found to promote hepatitis B and fibrosis by inhibiting solute carrier family 7a member 11-driven GPX4 expression and promoting ferroptosis in HSCs.[13] All the aforementioned mechanistic studies highlight the critical role of GPX4 as a downstream participant in the pathway, which modulates the initiation of ferroptosis and governs the progression of liver fibrosis.
We discovered several GPX4 co-expressed genes using the protein co-expression database. Among them, one notable finding was the co-expression coefficient of −0.5347 between GPX4 and the transcriptional repressor PR/SET domain 1 (PRDM1), with a P = 0.0039. PRDM1 is a repressor of interferon gene expression. The transcription of this gene increases upon virus induction.[14] Some studies have linked PRDM1 to liver-related diseases. Polymorphisms in the PRDM1 gene could influence HCC progression and prognosis.[15] Moreover, higher levels of PRDM1 in HCC patients have been associated with poorer prognosis and increased mortality rates.[16] Similarly, PRDM1 has been associated with prognosis in chronic HBV-infected diseases and HBV-associated HCC.[16] PRDM1 has been shown to promote ferroptosis in thyroid cancer by regulating the ubiquitin-specific peptidase 15 -mediated deubiquitination of selenium-binding protein 1.[17] However, the specific mechanism through which PRDM1 contributes to hepatitis B fibrosis is still unclear.
This investigation aimed to explore the impact of PRDM1 on HSC activity, hepatocyte ferroptosis, and its interaction with GPX4 to clarify the mechanism behind the involvement of PRDM1 in hepatitis B fibrosis and identify potential novel treatment direction for hepatic fibrosis.
MATERIAL AND METHODS
Organization sample collection
Tissue samples and clinical data were collected from 90 patients with CHB. They were diagnosed by liver tissue biopsy and admitted to our hospital between 2020 and 2021. Based on the Ishak scoring system, three groups of liver tissue samples were classified according to pathological staging: mild hepatic fibrosis (Ishak = S1), moderate hepatic fibrosis (Ishak = S2), and severe liver fibrosis (Ishak = S3–S4). The obtained liver tissue specimens were immediately stored in liquid nitrogen.
RNA extraction
A total of 50 mg of liver tissue was collected, and 500 μL of NucleoZol (740404.200, Gene Biotechnology International Trade, Shanghai, China) was added. The tissue was crushed at a low temperature using a tissue crusher (Homogenizer, Servicebio, Wuhan, China). The mixture was allowed to sit at room temperature for 15 min, with mixing and digestion occurring three to four times during this period. A total of 200 μL of enzyme-free water (BL510B, Biosharp, Anhui, China) were added, and the mixture was shaken vigorously for 15 s at a speed of 12,000 g. This procedure was followed by centrifugation at room temperature for 15 min. The supernatant was transferred to an EP tube. An equal amount of isopropyl alcohol (67-63-0, Sinopharm, Shanghai, China) was added and mixed. The mixture was left to stand for 10 min at room temperature before being centrifuged at a speed of 12,000 g for 10 min. The supernatant was removed, and 500 μL of 75% ethanol was added (64-17-5, Sinopharm, Shanghai, China). The RNA precipitate was washed by gently moving the tube up and down several times. Centrifugation was conducted for 3 min at a speed of 8,000 g. The washing step was repeated once more. Any remaining ethanol was removed and discarded, and the lid was opened to evaporate the ethanol for 2–5 min. Enzyme-free water was added to fully dissolve the RNA clusters, and the concentration was determined. The RNA was stored at −80°C.
Quantitative polymerase chain reaction
A total of 1 μg of RNA was collected, and reverse transcription was performed following the instructions provided in the Reverse Transcription Kit (G3333–50, Servicebio, Wuhan, China). An EP tube was prepared, and 10 μL of 2 × UltraSYBR Mixture (CW0957H, Kangwei Century Biotechnology, Jiangsu, China) was added. A total of 1 μL each of the reverse and forward primers, cDNA, and 7 μL of ddH2O were added. The reaction conditions were set to 95°C for 10 min for one cycle, 95°C for 15 s and 60°C for 1 min for 40 cycles, and a cycle of 15 s at 95°C and 15 s at 60°C. The experiment was repeated three times for each sample. The relative quantitative analysis method was used to analyze gene expression differences, with the formula as follows: ces = target gene of CT − reference gene of CT; ces, three times for each sample. The relative quantitative = 2-ΔΔCT. The primers used were as follows: PRDM1 forward primer: GACGAAGCGAGGAGGGAC; PRDM1 reverse primer: TCCCTGAAACCTCACAGTGC; GPX4 forward primer: GGAGCCAGGGAGTAACGAAG; GPX4 reverse primer: GACGGTGTCCAAACTTGGTG; GAPDH forward primer: CGGGAAGGA AATGAATGGGC; and GAPDH reverse primer: GGAAAAGCATCACCCGGAGG.
Cell culture
The RPMI 1640 basic culture medium (E600028, BBI, Shanghai, China) was prepared as a complete culture medium, with 10% serum (FSD500, Excell Bio, Jiangsu, China) and 100 U/mL each of penicillin and streptomycin (BL505A, Biosharp, Hefei, China). The culture medium was added to cultivate LX-2 (CL-0560, Punosai Life Science and Technology, Wuhan, China). The LX-2 cells were tested for mycoplasma and identified by short tandem repeat, which showed no mycoplasma contamination or cross-contamination with other human cells. It confirmed that the identity of the cell line was consistent with the source cell bank. The LX-2 cells were cultured at 37°C and with 5% CO2 in a cell culture incubator (Forma Direct Heat CO2 Incubators, Thermo Fisher Scientific, Massachusetts, USA) until reaching 70–80% confluence. Subsequently, the cells were digested with trypsin (25200072, Gibco, Grand Island, USA), counted with 106 cells per well, and inoculated into 6-well plates.
Cell transfection
Cells were inoculated into 6-well plates and cultured in an incubator. The culture medium was removed and replaced with a complete medium without double antibiotic. EP tubes A and B were prepared by adding 125 μL opti-minimum essential medium (31985070, Gibco, Grand Island, USA) to both tubes. Subsequently, appropriate amounts of plasmid and lipo3000 (L3000075, Invitrogen, California, USA) were added to tube A and mixed thoroughly. Meanwhile, 5 μL Lipo3000 was added to tube B and mixed thoroughly. Tubes A and B were left to stand for 5 min and then mixed together, followed by a 30 min incubation. Subsequently, the solution was added dropwise into the six-well plate and incubated for 6 h. After removing the original medium and adding the complete medium, the overexpression efficiency was verified after 48 h.
Cellular ROS activity assay
Following the instructions of the ROS Activity Assay Kit (S0035S, Beyotime Biotechnology, Shanghai, China), a serum-free medium was used to dilute CM-H2DCFDA to 5 μm/L. The cells were then harvested cells and resuspended in CM-H2DCFDA at a concentration of 107–2 × 108/mL at 37°C for 20 min, with mixing by inversion every 3–5 min. After washing the cells three times with serum-free medium, the cell fluorescence intensity was detected at 525 nm using a flow cytometer (DxFLEX, Beckman Coulter, California, USA).
Cellular malondialdehyde (MDA) concentration assay
Cells were lysed using cell lysate (P0013B, Beyotime Biotechnology, Shanghai, China). A volume of 0.1 mL of lysate was used for every 107 cells and kept at 0°C–4°C for half an hour. The mixture was then centrifuged at 12,000 g for 10 min, and the supernatant concentration was determined. The MDA concentration in cells was detected using the MDA detection kit (BC0025, Solarbio, Beijing, China). Two EP tubes were prepared. Notably, 300 μL of MDA detection working solution, 100 μL of sample, and 100 μL of Reagent No. 3 were added to the measurement tube and mixed well. Then, 300 μL of MDA detection working solution, 100 μL of Deuterium Depleted Water, and 100 μL of Reagent No. 3 were added to the blank tube and mixed well. The mixed solution was incubated at 100°C in a water bath for 60 min and then placed in an ice bath to cool. The solution was centrifuged at 10,000 g at room temperature for 10 min. A total of 200 mL of the supernatant was removed into a 96-well plate, and the absorbance of each sample was measured at 532 and 600 nm using a microplate reader (SpectraMax i3X, Molecular Devices, Shanghai, China). The calculations were performed as follows: ΔA532 = A532✶−A532, ΔA600 = A600−A600 blank, and ΔA = ΔA532−ΔA600.
Cell proliferative capacity assay
A total of 2,000 cells were added into each well of a 96-well plate, with wells designated for sampling at 0, 24, 48, and 72 h. The seeded plate was placed back into the incubator for 6 h. Therefore, 10 μL of MTT (E606334–0500, BBI, Shanghai, China) was introduced to the 0 h sample wells and incubated in the cell culture incubator for 1.5 h. Subsequently, the OD value was detected at 450 nm using a microplate reader (SpectraMax i3X, Molecular Devices, Shanghai, China). This process was repeated for the remaining wells at 24, 48, and 72 h sequentially.
Protein extraction and western blot analysis
Cells were collected and lysed in a 100 μL lysate mixture supplemented with the protease inhibitor phenylmethanesulfonylfluoride (ST2573–5 g, Beyotime Biotechnology, Shanghai, China). The cell suspension was kept on ice for 30 min with intermittent mixing. It was then centrifuged at 4°C and 12,000 g for 15 min. The supernatant was carefully aspirated and subsequently measured using a BCA kit (P0010S, Beyotime Biotechnology, Shanghai, China). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was conducted on a 10% acrylamide gel, with equal amounts (20 μg) of protein loaded onto each group. Following electrophoresis, proteins were transferred from the gel to a 0.45 μm polyvinylidene fluoride membrane (IPVH, Millipore, Massachusetts, USA) at a current of 300 mA for 1 h. The membranes were sealed in 5% skim milk (1172, BioFroxx, Guangdong, China) and shaken horizontally for 1 h. The membranes were incubated at 4°C in the laboratory overnight. Thereafter, the membranes were incubated with 0.55 μg/mL GPX4 (30388-1-AP, Proteintech, Wuhan, China), 0.2 μg/mL PRDM1 (61167, Proteintech, Wuhan, China), 0.6 μg/mL a-smooth muscle actin (a-SMA, 14395-1-AP, Proteintech, Wuhan, China), 0.48 μg/mL vimentin (22031-1-AP, Proteintech, Wuhan, China), 0.035 μg/mL desmin (16520-1-AP, Proteintech, Wuhan, China), and 0.05 μg/mL b-actin (66009-1-Ig, Proteintech, Wuhan, China) antibodies at laboratory temperature. TBS buffer containing 2‰ Tween 20 (9005-64-5, BBI, Shanghai, China) was prepared and used to rinse the membrane three times, each for 5 min. Thereafter, the membranes were incubated in 0.08 μg/mL Goat Anti-Mouse immunoglobulin G (IgG) (H+L)-HRP Conjugate (31430, Thermo Fisher Scientific, Massachusetts, USA) and 0.08 μg/mL Goat Anti-Rabbit IgG (H+L)-HRP Conjugate (31460, Thermo Fisher Scientific, Massachusetts, USA) antibodies. A low Fekker class ultra-sensitive ECL luminescent solution (MA0187-3, MeilunBio, Dalian, China) was added to the membrane, which was incubated for 30 s in the dark and photographed using an intelligent fluorescence chemiluminescence imaging system (GBOXCHE MIXRQ, Sygene, Cambridge, USA).
Statistical analysis
Research data were analyzed using the Statistical Package for the Social Sciences 20.0 (20.0, IBM, New York, USA) and expressed as mean ± standard deviation (s). For count data, the c2 test was employed for two-way unordered variables or when only grouped variables were ordered. On the contrary, the rank sum test was utilized when only response variables were ordered. For measurement data, an independent sample t-test was conducted for two-group comparisons. Meanwhile, analysis of variance was employed for multi-group comparisons. Non-parametric tests were used when the data did not meet the assumption of normality. A P < 0.05 indicated statistical significance.
RESULTS
GPX4 is negatively correlated with PRDM1
GPX4, as a key regulatory factor of ferroptosis, plays a significant role in its mechanism. Therefore, we identified a series of co-expressed genes of GPX4 through the Liver Disease Transcriptome Database at https://seek.princeton.edu. We selected the top 30 genes that showed a significant negative correlation with GPX4 and found that PRDM1 is a transcriptional repressor that plays an important role in gene regulation. The co-expression coefficient between PRDM1 and GPX4 was −0.5347, with a P = 0.0039. Therefore, it could potentially be a key gene in the ferroptosis pathway [Table 1].
| Gene | Entrez ID | Co-expression Score | P-value | Description |
|---|---|---|---|---|
| TSHZ3 | 57616 | −0.6514 | 0.0025 | Teashirt zinc finger homeobox 3 |
| SLC8A1 | 6546 | −0.6187 | 0.0023 | Solute carrier family 8 (sodium/calcium exchanger), member 1 |
| PAN3 | 255967 | −0.5973 | 0.0028 | PAN3 poly(A) specific ribonuclease subunit homolog (S. cerevisiae) |
| INTS6 | 26512 | −0.595 | 0.0043 | Integrator complex subunit 6 |
| RPGR | 6103 | −0.5905 | 0.0023 | Retinitis pigmentosa GTPase regulator |
| KIAA2026 | 158358 | −0.581 | 0.003 | KIAA2026 |
| RASA2 | 5922 | −0.5714 | 0.0033 | RAS p21 protein activator 2 |
| DNAJC18 | 202052 | −0.57 | 0.0024 | DnaJ (Hsp40) homolog, subfamily C, member 18 |
| MON2 | 23041 | −0.5639 | 0.0035 | MON2 homolog (S. cerevisiae) |
| BCL11B | 64919 | −0.5634 | 0.0025 | B-cell CLL/lymphoma 11B (zinc finger protein) |
| MTMR9LP | 339483 | −0.5621 | 0.0029 | Myotubularin related protein 9-like, pseudogene |
| AMPD1 | 270 | −0.5595 | 0.0023 | Adenosine monophosphate deaminase 1 |
| XYLT1 | 64131 | −0.5577 | 0.0035 | Xylosyltransferase I |
| CHD2 | 1106 | −0.5567 | 0.0041 | Chromodomain helicase DNA binding protein 2 |
| ZSWIM6 | 57688 | −0.5532 | 0.0031 | Inc finger, SWIM-type containing 6 |
| MIAT | 440823 | −0.5517 | 0.0039 | Myocardial infarction associated transcript (non-protein coding) |
| VCAN | 1462 | −0.5487 | 0.0031 | Versican |
| MED13L | 23389 | −0.5485 | 0.0031 | Mediator complex subunit 13-like |
| ZNF469 | 84627 | −0.5479 | 0.0033 | Zinc finger protein 469 |
| CHD1 | 1105 | −0.5471 | 0.0075 | Chromodomain helicase DNA binding protein 1 |
| AAK1 | 22848 | −0.5464 | 0.0039 | AP2 associated kinase 1 |
| LAYN | 143903 | −0.546 | 0.0036 | Layilin |
| ATAD2B | 54454 | −0.5446 | 0.0044 | ATPase family, AAA domain containing 2B |
| RIMKLB | 57494 | −0.5425 | 0.0028 | Ribosomal modification protein rimK-like family member B |
| KIAA1324L | 222223 | −0.5371 | 0.0028 | KIAA1324-like |
| PCM1 | 5108 | −0.5369 | 0.0027 | Pericentriolar material 1 |
| ZMYM2 | 7750 | −0.5364 | 0.004 | Zinc finger, MYM-type 2 |
| PRDM1 | 639 | −0.5347 | 0.0039 | PR domain containing 1, with ZNF domain |
| BASP1 | 10409 | −0.5335 | 0.0026 | Brain abundant, membrane attached signal protein 1 |
| NRG2 | 9542 | −0.5314 | 0.0013 | Neuregulin 2 |
The red letters display the data for PRDM1.
GPX4 and PRDM1 are closely linked to the development and progression of liver fibrosis
Tissues exhibiting varying degrees of hepatic fibrosis were collected to explore the association between PRDM1 and hepatic fibrosis and establish the relationship between GPX4 and hepatic fibrosis. These tissues were categorized into three stages: S1, S2, and S3–S4. Each stage comprised 30 cases. The expression of PRDM1 and GPX4 in fibrotic liver tissues of each group was detected by quantitative polymerase chain reaction. Our findings showed a remarkable increase in the messenger ribonucleic acid (mRNA) levels of GPX4 with the worsening of hepatic fibrosis, whereas PRDM1 exhibited an opposite pattern [Figure 1].
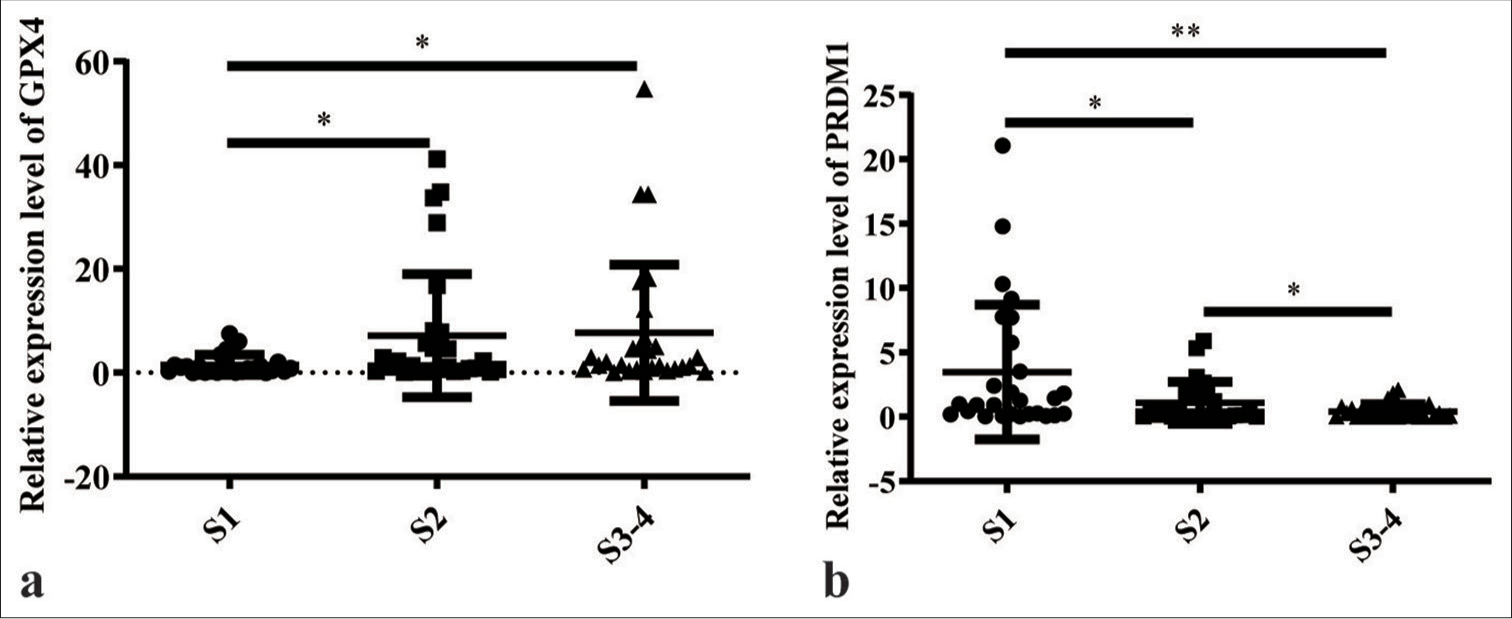
- Messenger ribonucleic acid (mRNA) levels of GPX4 and PRDM1 in different degrees of liver fibrosis. (a) mRNA levels of GPX4 in liver tissues with different degrees of hepatic fibrosis. (b) mRNA levels of PRDM1 in liver tissues with different degrees of hepatic fibrosis. S1: Mild fibrosis; S2: Moderate fibrosis; S3–S4: Advanced fibrosis and above; ✶P<0.05; ✶✶P<0.01. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4.
PRDM1 inhibits the activation of HSCs and facilitates their ferroptosis
Interfering and overexpressing PRDM1 cell lines were generated to elucidate the mechanism of PRDM1 in liver fibrosis. Furthermore, the expression of PRDM1 protein was assessed to confirm the successful establishment of the cell lines. Moreover, we evaluated the levels of ROS activity, MDA concentration, and cell proliferation capacity, as well as GPX4 and activation markers a-SMA, vimentin, and desmin, in HSCs. The findings revealed that overexpressing PRDM1 cell lines were successfully established [Figure 2a and b]. In addition, the protein level of GPX4 was hampered by the overexpression of PRDM1 [Figure 2c]. The upregulation of PRDM1 significantly diminished the levels of cellular activation marker proteins in HSCs [Figure 2d-f]. Similarly, interfering PRDM1 cell lines were successfully established [Figure 2g and h], whereas the protein level of GPX4 was significantly enhanced after PRDM1 interference [Figure 2i], interference with PRDM1 led to a significant increase in the levels of cellular activation marker proteins in HSCs [Figure 2j-l]. The overexpression of PRDM1 in HSCs led to a substantial elevation in ROS activity and MDA concentration, which were markedly suppressed upon interference with PRDM1 [Figure 3a-c]. Moreover, the overexpression of PRDM1 substantially reduced the cellular activity of HSCs, whereas interference with PRDM1 significantly increased it [Figure 3d].
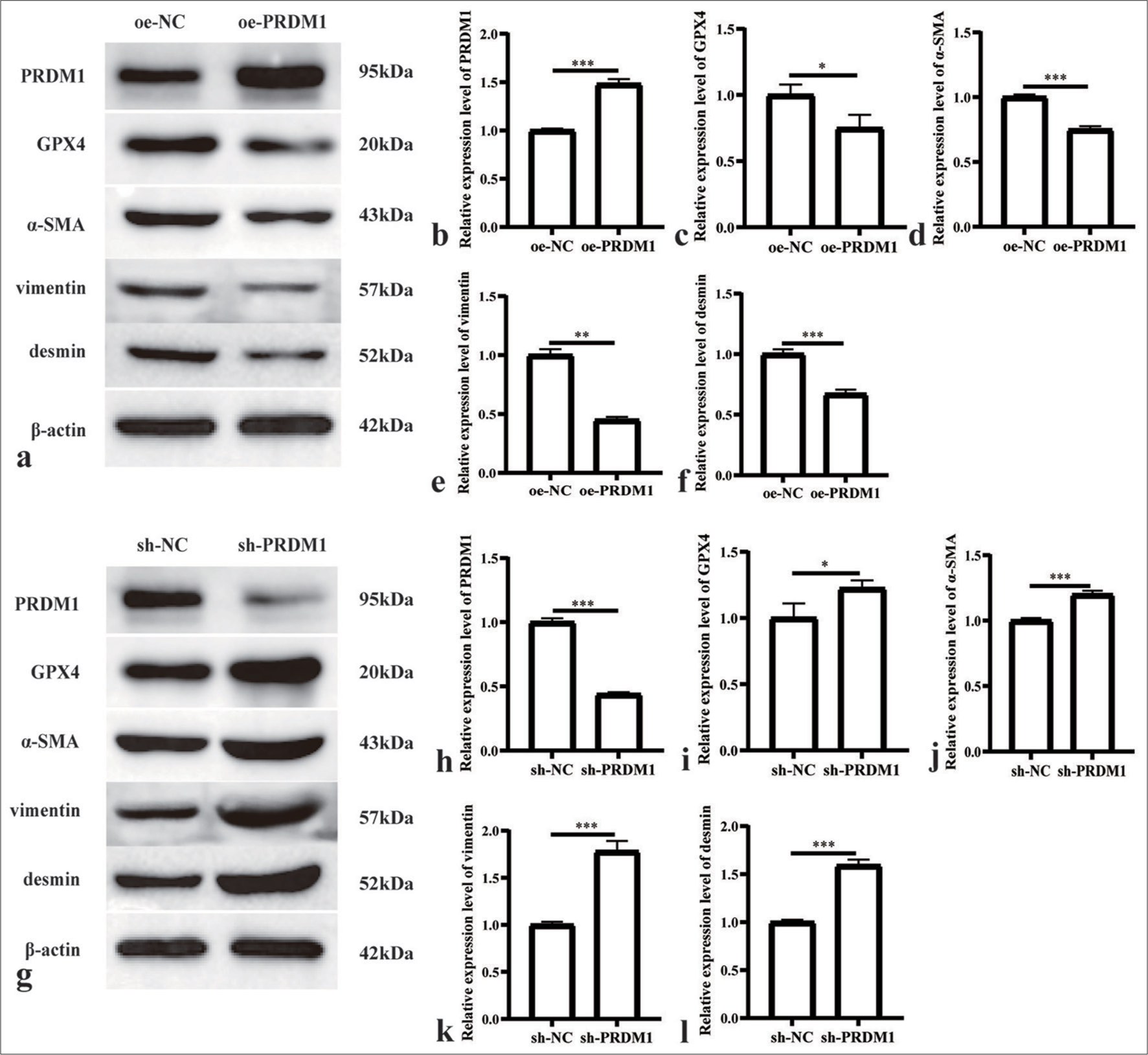
- Overexpression of PRDM1 inhibits the activity of HSCs. (a) Western blot plots of PRDM1, GPX4, and HSC cell activation marker proteins alpha smooth muscle actin (α-SMA), vimentin, and desmin in HSCs of the overexpression control (oe-NC) and PRDM1 overexpression (oe-PRDM1) groups. (b-f) Expression of the abovementioned proteins in oe-NC and oe-PRDM1 groups. (g) Western blot plots of PRDM1, GPX4, a-SMA, vimentin, and desmin in HSCs of the interfering control (sh-NC), and PRDM1 interfering (sh-PRDM1) groups. (h-l) Expression of the abovementioned proteins in sh-NC and sh-PRDM1 groups. ✶P<0.05; ✶✶P<0.01; ✶✶✶P<0.001. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, ROS: Reactive oxygen species, HSC: Hepatic stellate cells.
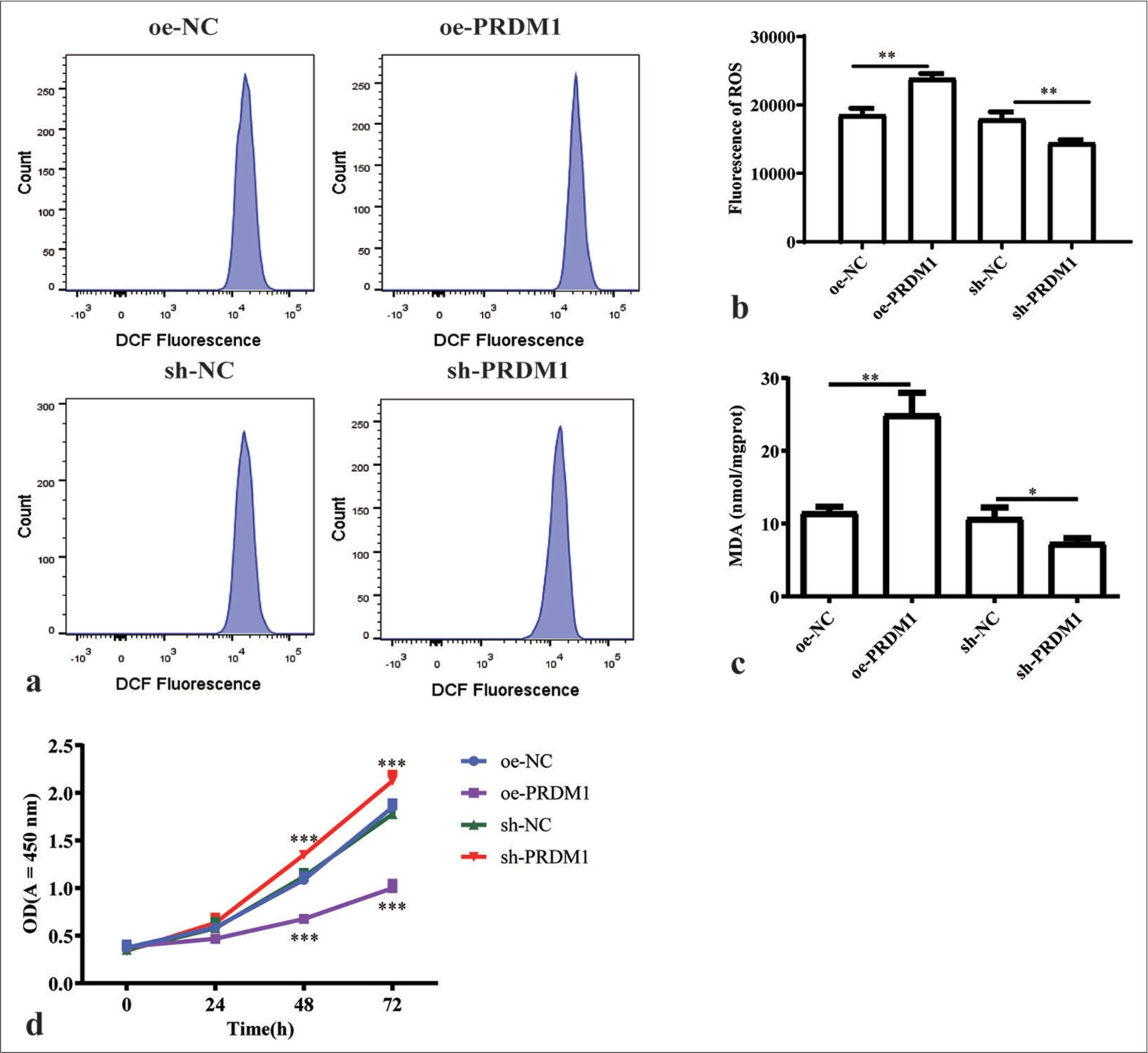
- Overexpression of PRDM1 promotes ferroptosis and inhibits the proliferation of HSCs. (a) Fluorescence intensity flow diagram of ROS in HSCs of the overexpression control (oe-NC), PRDM1 overexpression (oe-PRDM1), interfering control (sh-NC), and PRDM1 interfering (sh-PRDM1) groups. (b) Quantification of the fluorescence intensity of ROS. (c) Concentration of malondialdehyde in the four groups of cells mentioned above. (d) Proliferation of cells in the four groups of cells mentioned above at 0, 24, 48, and 72 h. ✶P<0.05; ✶✶P<0.01; ✶✶✶P<0.001. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, ROS: Reactive oxygen species, HSC: Hepatic stellate cells, DCF: Dichlorofluorescein, sh-NC: Negative Control, sh-RNA: Short hairpin RNA.
PRDM1 functions as an upstream regulator of GPX4 in the cellular context
We generated cell lines with overexpressed and knocked-down GPX4 to investigate the upstream and downstream association between PRDM1 and GPX4. The expression levels of GPX4 and PRDM1 were detected by Western blot analysis. The results revealed that altering the expression level of GPX4 had no discernible impact on the expression level of PRDM1 [Figure 4a-c]. At the same time, we detected GPX4 mRNA levels in cell lines with overexpressed and knocked down PRDM1. Moreover, GPX4 mRNA levels decreased significantly with the overexpression of PRDM1 and increased significantly with the knockdown PRDM1 [Figure 4d].
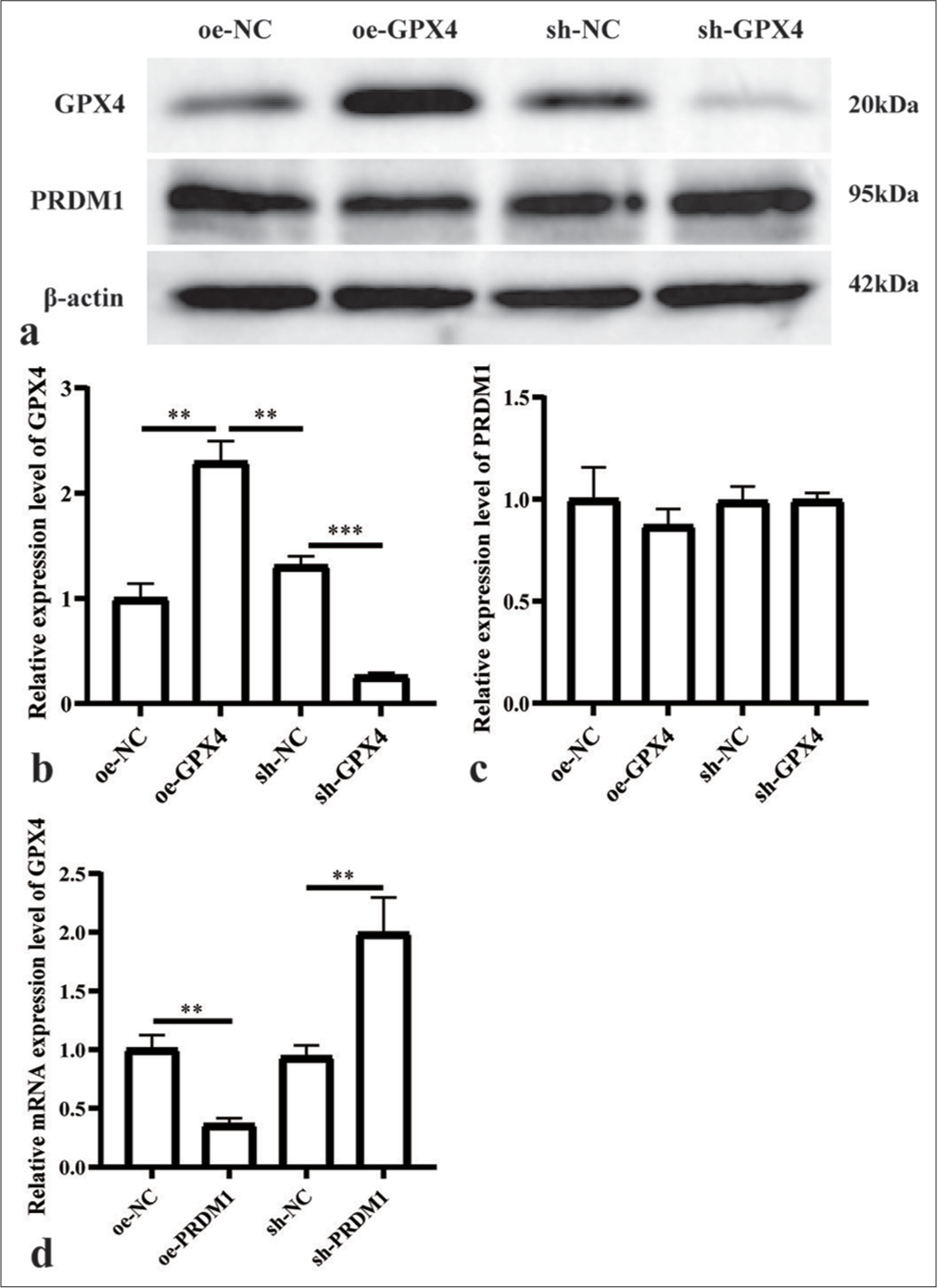
- PRDM1 is the upstream regulator of GPX4. (a) Western blot results for PRDM1 and GPX4 proteins; (b and c) Expression of the abovementioned proteins in different cell groups. (d) GPX4 mRNA levels in cell lines with overexpressed and knocked down PRDM1. ✶✶P<0.01; ✶✶✶P<0.001. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, mRNA: Messenger ribonucleic acid, sh-NC: Negative Control, sh-RNA: Short hairpin RNA.
PRDM1 regulates GPX4-induced ferroptosis to impact the expression of HSC activation markers
We genetically modified HSCs to generate various cell lines for investigating the mechanism of PRDM1/GPX4 in ferroptosis in liver fibrosis. These cell lines included shNC, sh-PRDM1, sh-PRDM1 + sh-GPX4, sh-PRDM1 + sh-GPX4 + Fer-1, oe-NC, oe-PRDM1, oe-PRDM1 + oeGPX4, and oe-PRDM1 + oe-GPX4 + Erastin. We then examined the ROS activity, MDA concentration, cell proliferation, and expression levels of GPX4, as well as the cell activation marker proteins (a-SMA, vimentin, and desmin), in each group of HSCs. Our findings demonstrated that simultaneous interference with PRDM1 and GPX4 resulted in increased ROS activity and MDA concentration compared with interference with PRDM1 alone. When the ferroptosis inhibitor (Fer-1) was added, a trend of decreasing ROS activity and MDA concentration was observed [Figures 5a-c]. Compared with interfering only with PRDM1, simultaneous interference with PRDM1 and GPX4 resulted in a decrease in cell proliferation ability, which was further enhanced upon the addition of a Fer-1. [Figure 5d]. Conversely, simultaneous overexpression of PRDM1 and GPX4 decreased ROS activity and MDA concentration compared with the overexpression of PRDM1 alone. When the ferroptosis activator (Erastin) was added, a trend of elevating ROS activity and MDA concentration was found [Figures 6a-c], compared with interfering only with PRDM1, the overexpression of PRDM1 and GPX4 led to an increase in cell proliferation ability, which was attenuated upon the addition of a ferroptosis activator [Figures 6d].
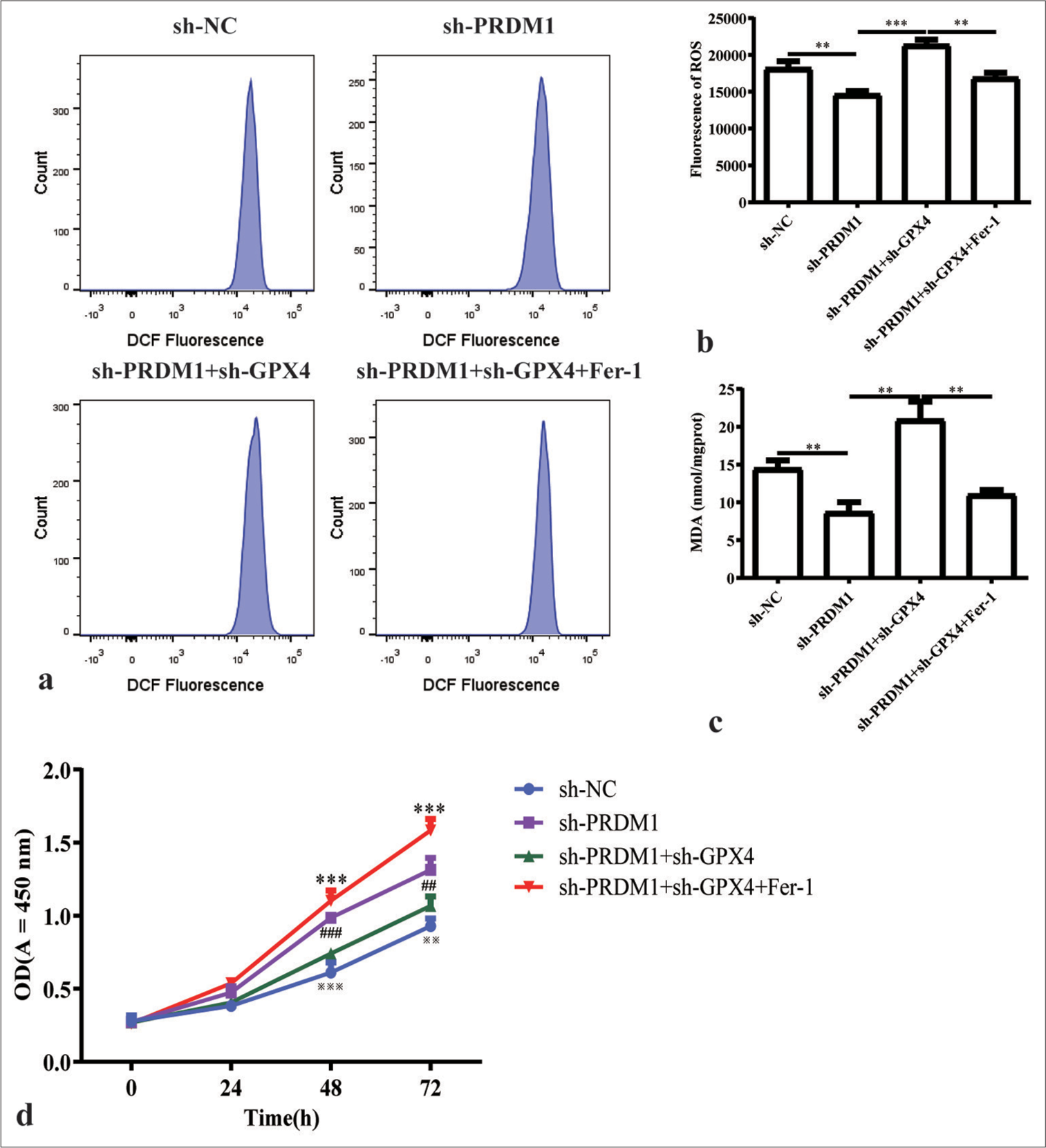
- Simultaneous silencing of PRDM1 and GPX4 promotes ferroptosis and inhibits the proliferation of HSCs. (a) Fluorescence intensity flow pattern of ROS in liver stellate cells in the sh-NC, sh-PRDM1, sh-PRDM1+sh-GPX4, and sh-PRDM1+sh-GPX4+Fer-1 groups. (b) Quantification of the fluorescence intensity of ROS in the four groups of cells mentioned above. (c) Concentration of malondialdehyde in HSCs in the four groups of cells mentioned above; Fer-1 is a ferroptosis inhibitor. (d) Cell proliferation of HSCs in the four groups of cells mentioned above at 0, 24, 48, and 72 h. ✶: sh-PRDM1 + sh-GPX4 vertus sh-PRDM1 + sh-GPX4 + Fer-1; #: shPRDM1 versus sh-PRDMA + sh-GPX4; ※: sh-NC versus sh-PRDM1; ✶✶✶P<0.001; ##P<0.01, ###P<0.001; ※※P<0.01, ※※※P<0.001. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, ROS: Reactive oxygen species, HSC: Hepatic stellate cells, a-SMA: a-smooth muscle actin, Fer-1: Ferroptosis inhibitor, DCF: Dichlorofluorescein, sh-NC: Negative Control, sh-RNA: Short hairpin RNA.
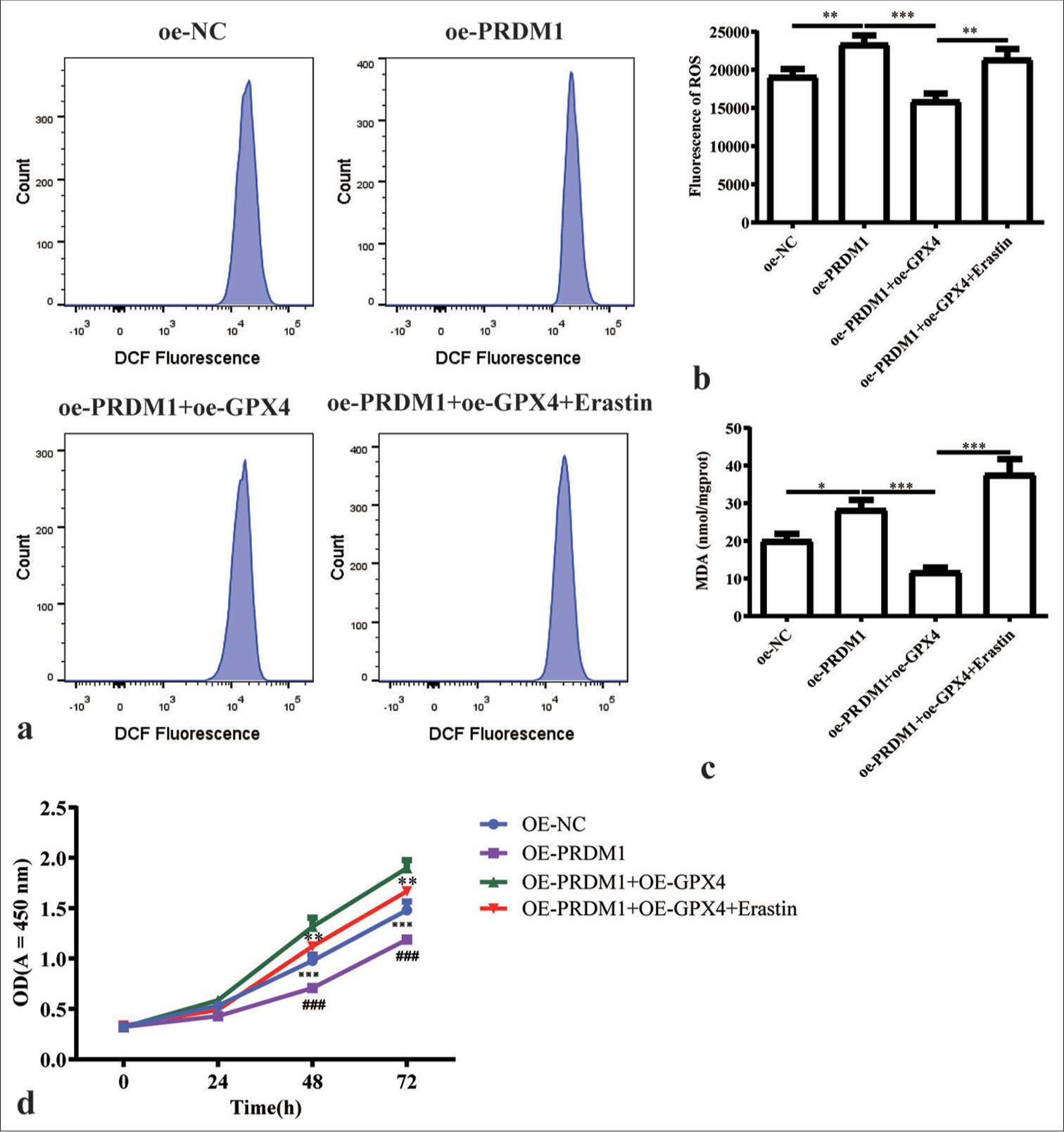
- Simultaneous overexpression of PRDM1 and GPX4 promotes ferroptosis and inhibits the proliferation of HSCs. (a) Fluorescence intensity of ROS in cells in the oe-NC, oe-PRDM1, oe-PRDM1 + oe-GPX4, and oe-PRDM1 + oe-GPX4 + Erastin groups of cells in the fluorescence intensity flow diagram; Erastin is a ferroptosis inducer. (b) Quantification of the fluorescence intensity of ROS in the four groups of cells mentioned above. (c) Concentration of malondialdehyde in HSCs in the four groups of cells mentioned above; Fer-1 is a ferroptosis inhibitor. (d) Cell proliferation of HSCs in the four groups of cells mentioned above at 0, 24, 48, and 72 h. ✶: OE-PRDM1 + OE-GPX4 versus OE-PRDM1 + OE-GPX4 + Erastin; #: OE-PRDM1 versus OE-PRDMA + OE-GPX4; ※: OE-NC versus OE-PRDM1; ✶✶P<0.01, ✶✶✶P<0.001; ###P<0.001; ※※※P<0.001. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, ROS: Reactive oxygen species, HSC: Hepatic stellate cells, a-SMA: a-smooth muscle actin, Fer-1: Ferroptosis inhibitor, DCF: Dichlorofluorescein, oe-NC: Overexpressing negative control.
Notably, the levels of HSC activation markers were significantly reduced upon interference with PRDM1 and GPX4. However, they were markedly increased upon supplementation with a Fer-1. Conversely, these markers were increased remarkably when PRDM1 and GPX4 were overexpressed, but they were notably decreased upon the addition of a ferroptosis activator [Figures 7 and 8].
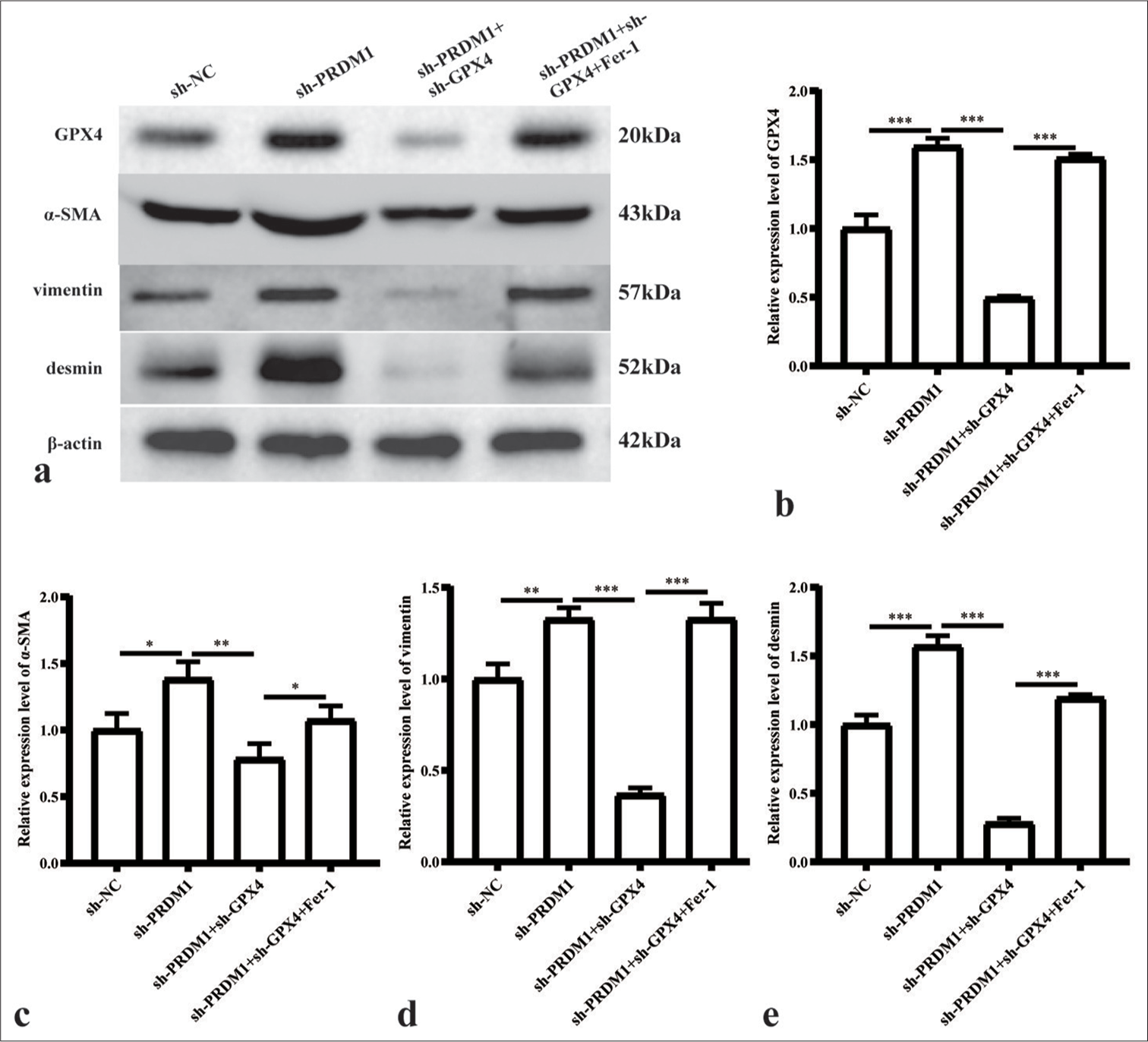
- Simultaneous silencing of PRDM1 and GPX4 inhibits the expression of active proteins in HSCs. (a) Western blot plots of GPX4 and HSC cell activation marker proteins a-SMA, vimentin, and desmin in HSCs of the sh-NC, sh-PRDM1, sh-PRDM1 + sh-GPX4, and sh-PRDM1 + sh-GPX4 + Fer-1 groups. (b-e) Statistical graphs of the expression of the abovementioned proteins in different cell groups. ✶: P<0.05; ✶✶: P<0.01; ✶✶✶: P<0.001 for the expression of the abovementioned proteins. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, HSC: Hepatic stellate cells, a-SMA: a-smooth muscle actin, Fer-1: Ferroptosis inhibitor, sh-NC: Negative Control, sh-RNA: Short hairpin RNA.
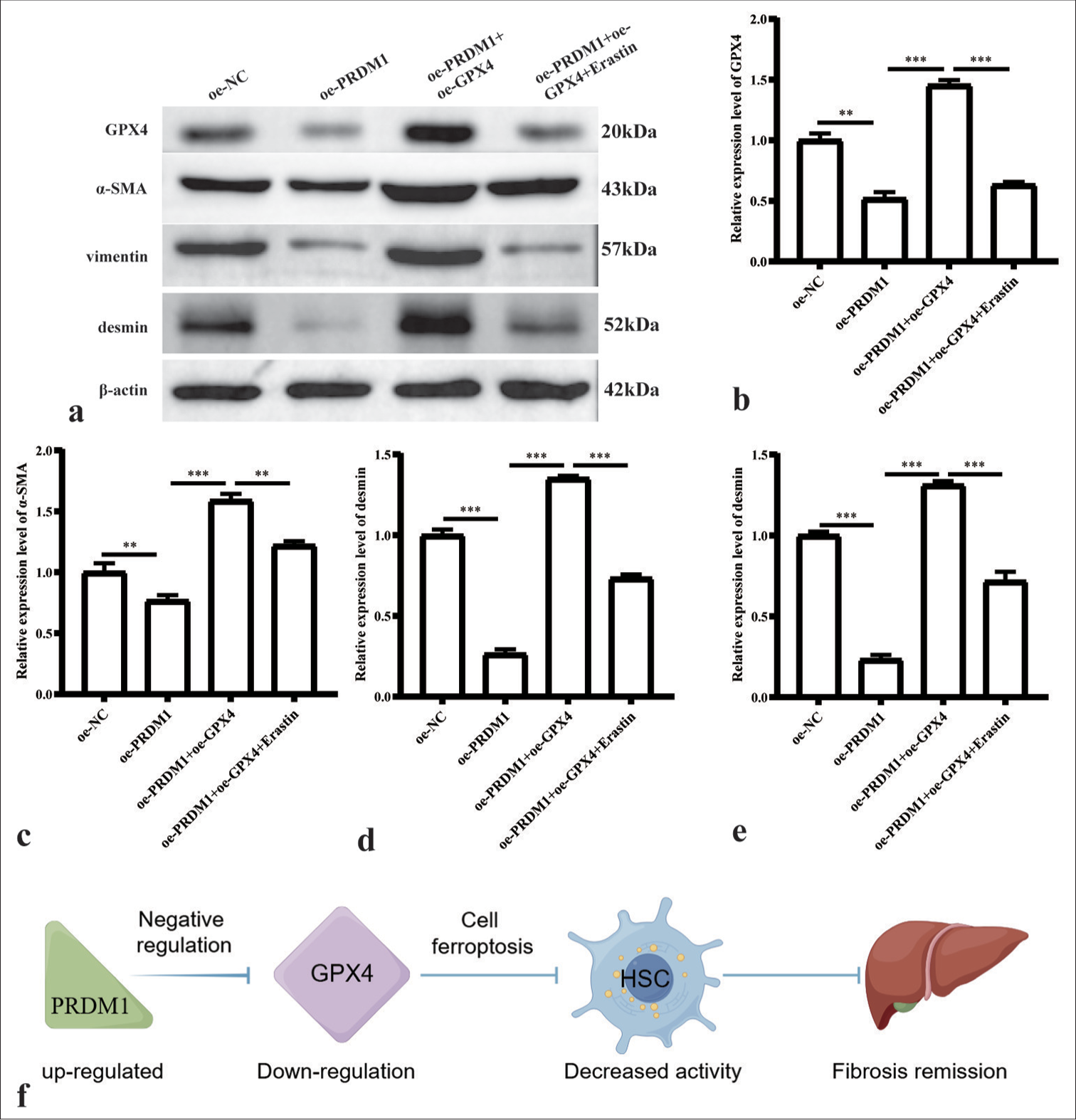
- Simultaneous overexpression of PRDM1 and GPX4 inhibits the expression of active proteins in HSCs. (a) Western blot plots of GPX4 and HSC cell activation marker proteins a-SMA, vimentin, and desmin in the oe-NC, oe-PRDM1, oe-PRDM1+oe-GPX4, and oe-PRDM1+oe-GPX4+Erastin groups. (b-e) Statistical graph of the expression of the abovementioned proteins in different cells. ✶✶: P<0.01; ✶✶✶: P<0.001. (f) Pathway of PRDM1-GPX4 in HSCs. Each experiment was repeated three times. PRDM1: PR/SET domain 1, GPX4: Glutathione peroxidase 4, HSC: Hepatic stellate cells, a-SMA: a-smooth muscle actin.
DISCUSSION
HSCs are considered the main driver of hepatic fibrosis. Notably, the regression of liver fibrosis is associated with a decline or loss in activated HSCs. Therefore, inactivating or reducing HSCs could serve as a potential anti-fibrotic strategy.[18] HSCs are rich in iron, which is essential for ferroptosis.[19] Some studies have demonstrated that inducing ferroptosis in HSCs can effectively reverse liver fibrosis.[20] The enzyme GPX4 is a key regulatory factor in the cellular ferroptosis mechanism. A deficiency in GPX4 reduces cellular antioxidant capacity, which results in the accumulation of ROS and subsequently triggers ferroptosis.[21] We investigated liver tissue samples displaying varying degrees of hepatic fibrosis and found a pronounced positive association between GPX4 level and the severity of fibrosis. This observation implies that tissues with more pronounced fibrosis display reduced occurrences of ferroptosis. To delve deeper into the underlying mechanisms governing ferroptosis in liver fibrosis, we utilized the Gene Interaction Database and discovered the transcriptional regulatory factor PRDM1 has a significant negative correlation with GPX4.
PRDM1 is important in maintaining and altering diverse cellular conditions. It has been implicated in chronic HBV infection and the prognosis of patients with HBV-associated HCC.[16] However, the role of PRDM1 in HBV-induced liver fibrosis remains unclear. We investigated the expression levels of PRDM1 in liver tissues with varying degrees of fibrosis. Remarkably, we observed a progressive decline in PRDM1 levels with increasing severity of hepatic fibrosis. Moreover, our findings demonstrated a negative correlation between the expression trends of PRDM1 and GPX4, which is consistent with our online prediction. HSC lines with PRDM1 overexpression and interference were generated to explore the function of PRDM1 on liver fibrosis. The overexpression of PRDM1 remarkably reduced the activation markers of HSC, namely a-SMA, vimentin, and desmin. These findings indicate that PRDM1 can effectively inhibit the activation of HSCs. Interfering with the expression of PRDM1 led to the opposite outcome. Inhibition of HSC activity has been shown to impede the progression and potentially reverse hepatic fibrosis,[22] which highlights the importance of PRDM1 expression in the development of liver fibrosis. Meanwhile, we investigated the influence of PRDM1 on ferroptosis in HSCs. Our results demonstrated that the expression of PRDM1 in these cells led to a significant increase in the concentrations of ROS and MDA, whereas the level of GPX4 protein was reduced. MDA serves as an indicator of lipid peroxidation and can be utilized to evaluate the occurrence of ferroptosis,[22] whereas GPX4 plays a crucial role in maintaining cellular redox homeostasis as an antioxidant enzyme.[4] These results indicate that enhanced expression of PRDM1 may promote ferroptosis in HSCs by suppressing GPX4 function.
We generated interference and overexpression cell lines for GPX4 to explore the association between PRDM1 and GPX4. Interestingly, altering the expression levels of GPX4 did not impact the protein levels of PRDM1. However, PRDM1 demonstrated the ability to suppress the protein expression of GPX4, which indicates its regulatory role upstream of GPX4. Subsequently, simultaneous interference of PRDM1 and GPX4 resulted in a notable inhibition of HSC activity and promotion of ferroptosis compared with cells with only PRDM1 interference. Importantly, the addition of a Fer-1 reversed the induction of ferroptosis and restored HSC activity, which highlights the pivotal role of GPX4 in ferroptosis induction, with PRDM1 acting as the regulatory protein. This finding is supported by the response experiment. Simultaneous overexpression of PRDM1 and GPX4 suggests that PRDM1 potentially triggers ferroptosis by modulating GPX4. In addition, we assessed the activation markers of HSCs across all groups, which revealed that PRDM1 predominantly regulates cellular ferroptosis by suppressing GPX4 gene transcription and thus promotes the activation of HSCs.
Although this study offers important insights into the mechanisms through which PRDM1-GPX4 regulates HSCs, some limitations need to be considered. First, the molecular mechanism has not been thoroughly explored, and experiments need to be designed to verify the direct targeting relationship between PRDM1 and GPX4. In addition, further investigation into more detailed regulatory elements in this pathway is needed to improve the understanding of the regulatory mechanism of ferroptosis. Second, due to time and funding constraints, this study was restricted to works at the cellular level in vitro, which did not fully reflect the regulatory effects of this pathway in humans. Future studies should incorporate animal experiments to validate the regulatory role of this pathway in vivo.
SUMMARY
We have uncovered a significant association between the transcriptional regulator PRDM1 and hepatic fibrosis. Our findings demonstrate that PRDM1 acts as an upstream regulator of GPX4, which exerts control over the initiation of cellular ferroptosis by inhibiting the transcription of GPX4. This mechanism ultimately promotes the activation of HSCs. The discovery of this novel pathway is crucial for future investigations into reversing the progression of liver fibrosis.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
ACKNOWLEDGMENT
Not applicable.
ABBREVIATIONS
17-OH PREG: 17α-hydroxypregnenolone
α-SMA: α-smooth muscle actin
BECN1: beclin 1
CHB: Chronic hepatitis B
GPR56: G protein-coupled receptor 56
GPX4: Glutathione peroxidase 4
GSH: Glutathione
HBF: Hepatitis B and Fibrosis
HBV: Hepatitis B virus
HCC: Hepatocellular carcinoma
HIF-1α: Hypoxia inducible factor-1α
HSCs: Hepatic stellate cells
MDA: Malondialdehyde
PRDI: Domain I element
PRDM1: PR/SET domain 1
ROS: Oxygen species
SELENBP1: Selenium-binding protein 1
SLC7A11: Solute Carrier Family 7 Member 11
USP15: Ubiquitin-specific peptidase 15
AUTHOR CONTRIBUTIONS
MHL and HBG: Conceptualization and supervision; HXM: Project administration and writing–review and editing; WJW and WHK: Writing-original draft; WPS: Validation and visualization; TL and SLL: Writing-original draft.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All the procedures were approved by the Medical Ethics Committee of Mengchao Hepatobiliary Hospital, Fujian Medical University (2021-061-01), dated 2021.06.08. All subjects signed written informed consent. This study follows the ethical principles of the Declaration of Helsinki.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
This work was supported by the Natural Science Foundation of Fujian Province (2021J011289 and 2021J011292) and Fuzhou Health Science Research Project for Middle and Young People (2021-S-wq24); Fuzhou Science and Technology Plan Project (2021-S-099); Fujian Provincial Clinical Medical Research Center for Infectious Diseases (2022Y2018).
References
- Natural regression of fibrosis in chronic hepatitis B. World J Gastroenterol. 2016;22:5459-66.
- [CrossRef] [PubMed] [Google Scholar]
- A novel prediction model for significant liver fibrosis in patients with chronic hepatitis B. Biomed Res Int. 2020;2020:6839137.
- [CrossRef] [PubMed] [Google Scholar]
- Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1a/SLC7A11 pathway. Cell Prolif. 2022;55:e13158.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151-66.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis: Past, present and future. Cell Death Dis. 2020;11:88.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72.
- [CrossRef] [PubMed] [Google Scholar]
- Ferroptosis: The good, the bad and the ugly. Cell Res. 2020;30:1061-2.
- [CrossRef] [PubMed] [Google Scholar]
- Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131.
- [CrossRef] [PubMed] [Google Scholar]
- Sensing steroid hormone 17a-hydroxypregnenolone by GPR56 enables protection from ferroptosis-induced liver injury. Cell Metab. 2024;36:2402-18.e10.
- [CrossRef] [PubMed] [Google Scholar]
- HucMSC-derived exosomes delivered BECN1 induces ferroptosis of hepatic stellate cells via regulating the xCT/GPX4 axis. Cell Death Dis. 2022;13:319.
- [CrossRef] [PubMed] [Google Scholar]
- Blimp1: A conserved transcriptional repressor critical for differentiation of many tissues. Exp Cell Res. 2009;315:1077-84.
- [CrossRef] [PubMed] [Google Scholar]
- The role of PRDM1 gene polymorphism in the progression of hepatocellular carcinoma in Egyptian patients. J Med Virol. 2023;95:e28343.
- [CrossRef] [PubMed] [Google Scholar]
- PRDM1 levels are associated with clinical diseases in chronic HBV infection and survival of patients with HBV-related hepatocellular carcinoma. Int Immunopharmacol. 2019;73:156-62.
- [CrossRef] [PubMed] [Google Scholar]
- PRDM1 promotes the ferroptosis and immune escape of thyroid cancer by regulating USP15-mediated SELENBP1 deubiquitination. J Endocrinol Invest. 2024;47:2981-97.
- [CrossRef] [PubMed] [Google Scholar]
- The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927-39.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083-103.
- [CrossRef] [PubMed] [Google Scholar]
- TUG1 protects against ferroptosis of hepatic stellate cells by upregulating PDK4-mediated glycolysis. Chem Biol Interact. 2023;383:110673.
- [CrossRef] [PubMed] [Google Scholar]
- Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31.
- [CrossRef] [PubMed] [Google Scholar]
- Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021;12:65.
- [CrossRef] [PubMed] [Google Scholar]




