


Translate this page into:
Serous fluids and hematolymphoid disorders

*Corresponding author: Ali Gabali, MD, PhD Professor, Director of Hematopathology and Hematopathology Fellowship, Department of Pathology, Wayne State University School of Medicine, Karmanos Cancer Center, Detroit, Michigan, United States. agabali@med.wayne.edu
-
Received: ,
Accepted: ,
How to cite this article: Gabali A. Serous fluids and hematolymphoid disorders. CytoJournal 2022;19:17.
HTML of this article is available FREE at: https://dx.doi.org/10.25259/CMAS_02_12_2021
Abstract
Diagnosing hematolymphoid neoplasm by evaluating fine-needle aspiration (FNA) cytology sample is controversial and requires experience and clinical skills. This concept becomes more challenging when evaluating hematolymphoid neoplasm in body fluid. Differentiating between low-grade lymphoma and reactive lymphocytes is often difficult by morphology alone as reactive lymphoid cells may acquire activation morphology from being exposed to different cytokines within the body fluid. However, in most cases there are specific features that may aid in differentiating small reactive from non-reactive lymphocytes including the round shape of the nucleus, the absence of visible nucleoli and the presence of fine clumped chromatin. In large cell lymphoma and leukemia cells involvement of body fluid this concept becomes less challenging. Large cell lymphoma and leukemia cells tend to have large size nuclei, less mature chromatin, and visible nucleoli with and without cytoplasmic vacuoles. However, to reach accurate diagnosis and subclassification, the utilizing of flow cytometry, to confirm monoclonality, and other ancillary studies such immunocytochemistry, cytogenetics and molecular studies is needed. This review article will be incorporated finally as one of the chapters in CMAS (CytoJournal Monograph/Atlas Series) #2. It is modified slightly from the chapter by the initial authors in the first edition of Diagnostic Cytopathology of Serous Fluids.
Keywords
Lymphoma
Leukemia
Small Cell
Large Cell
Body Fluid


INTRODUCTION
In general, hematolymphoid cells manifest as dyshesive cells in cytological specimens.[1] However, this is not a hard and fast rule, and exceptions can occur in some instances, either as a natural phenomenon or due to artifact. For example, it is known that lymphocytes in tuberculous effusions and in fine needle aspirations (FNAs) of follicular lymphoma may form ‘lymphoid aggregates’.[1,2] • An effusion-based lymphoma simulating carcinoma, even forming apparent ‘glands,’ and ‘papillary’ structures, has been described in the case of a pleural anaplastic large cell lymphoma.[3] In addition, cytoplasmic vacuoles are not uncommon in large cell lymphomas involving body cavities. Conversely, the cells of non-hematopoietic neoplasms, such as melanoma, neuroblastoma or desmoplastic small round cell tumor, can appear individually or in loose clusters mimicking lymphoma and leukemia cells.[4.5] Adding to the challenge, classic lymphoglandular bodies, representing remnants of lymphocyte cytoplasm, are typically inconspicuous or absent in effusion fluids, in contrast to FNA material from solid specimens.
After determining the lymphoid nature of the cells, the distinction between low-grade lymphoma and reactive lymphocytosis is often difficult by morphology alone, since cellular atypia is often minimal or absent in low-grade lymphoma. With high-grade lymphomas, the differential is usually between melanoma, neuroblastoma, undifferentiated sarcoma, and carcinoma. In most cases, an accurate diagnosis and classification can be achieved by integrating clinical history, cytologic findings, and immunophenotype with immunostains and/or flow cytometry [Table 1] along with molecular studies as indicated.
| Type of lymphoma | Cytomorphology | Figures (Diff-Quik stain) | Immunophenotype | Remarks | |
|---|---|---|---|---|---|
| B2a | Chronic lymphocytic leukemia / small lymphocytic lymphoma CLL/SLL | 1. Monomorphic, small, round, lymphocytes. 2. Clumped chromatin (“soccer-ball-like”). 3. Scant cytoplasm. 4. Indistinct or absent nucleoli. |
 |
1. Predominantly dim CD20 positive B-cells, without FMC7 2. Aberrantly expresses T-cell marker CD5 and usually also CD43. 3. Co-express CD23 but not cyclin-D1. |
In tissue, it shows varying numbers of prolymphocytes, paraimmunoblasts, and plasmacytoid lymphocytes that differ from the classically described small round lymphocyte (these features may be absent when involving body fluid). |
| B2b | Follicular lymphoma (grade 1-2) | 1. Monomorphic, small, irregular clefted nucleus (centrocytes) 2. Scant cytoplasm 3. Indistinct nucleoli |
 |
1. Express B-cell markers (CD20 and/or PAX-5) and CD10 2. Usually CD43 negative |
1. Higher grades of follicular lymphoma may have increased numbers of large lymphocytes (centroblasts) 2. CD10 cells also seen in follicular hyperplasia, but the germinal centers are bcl-2 negative |
| B2c | Burkitt lymphoma | 1. Monomorphic cells with medium sized nucleus 2. Scant basophilic cytoplasm with vacuoles |
 |
1. Similar immunophenotype to follicular lymphoma (i.e. CD10 positive) except usually lacks BCL-2 positivity 2. TdT negative |
Burkitt cells should be smaller than a macrophage nucleus. MIB-1 (KI67) should approach 100% and mitotic figures are readily seen |
| B2d | Diffuse large B-cell lymphoma (DLBCL) |
1. Pleomorphic population of cells with large nuclei 2. Single or multiple nucleoli 3. Moderate to abundant cytoplasm |
 |
1. Highly variable (May be CD10, CD5, BCL-2, or CD43 positive) 2. TdT negative |
This represents a heterogenous group that may develop de novo or from transformation of a low-grade lymphoma |
| B2e | Acute lymphoblastic leukemia / lymphoma (ALL/LBL) | 1. Small to medium sized round nucleus 2. Scant basophilic cytoplasm 3. Prominent nucleoli 4. Fine immature chromatin |
 |
1. Nuclear TdT positivity 2. Often CD43 positive 3. May be CD10 positive 4. May be T or B-cell phenotype |
Sometimes ALL cells may assume a “small” cell morphology and can mimic a low-grade lymphoma-leukemia like CLL/SLL |
Air-dried Diff-Quik (DQ) or Wright–Giemsa-stained slides have traditionally been the stains of choice for hematopoietic cells, especially for examining subtle cytoplasmic details and variations. However, alcohol-fixed, Papanicolaou (PAP)-stained preparations are generally better for examination of nuclear detail. In addition to preparing well stained smears and cell blocks[6-8] efforts should be made to save extra samples for ancillary studies, such as flow cytometry and molecular studies, when assessing possible hematopoietic neoplasms. The value of new cytologic preparations such as Thin-Prep has not been extensively evaluated in examining effusion cytology, particularly regarding hematopoietic cells.
Epidemiology
In adults within the United States, pleural, peritoneal, and pericardial effusions are most commonly related to benign conditions like congestive heart failure, cirrhosis of the liver, and pericarditis. Malignant effusions are most commonly secondary to epithelial neoplasms (breast, lung, gastrointestinal, and genitourinary cancer). However, non-Hodgkin lymphoma accounts for approximately 10% of all malignant pleural effusions. After lung and breast carcinoma, lymphoma is the third most common cause of malignant pleural effusion.[9-12] However, in regards to malignant peritoneal effusions, lymphoma (7%) ranks third after ovary (32%) and breast (13%) carcinoma [see Table 2, Figure 3].[9,10]
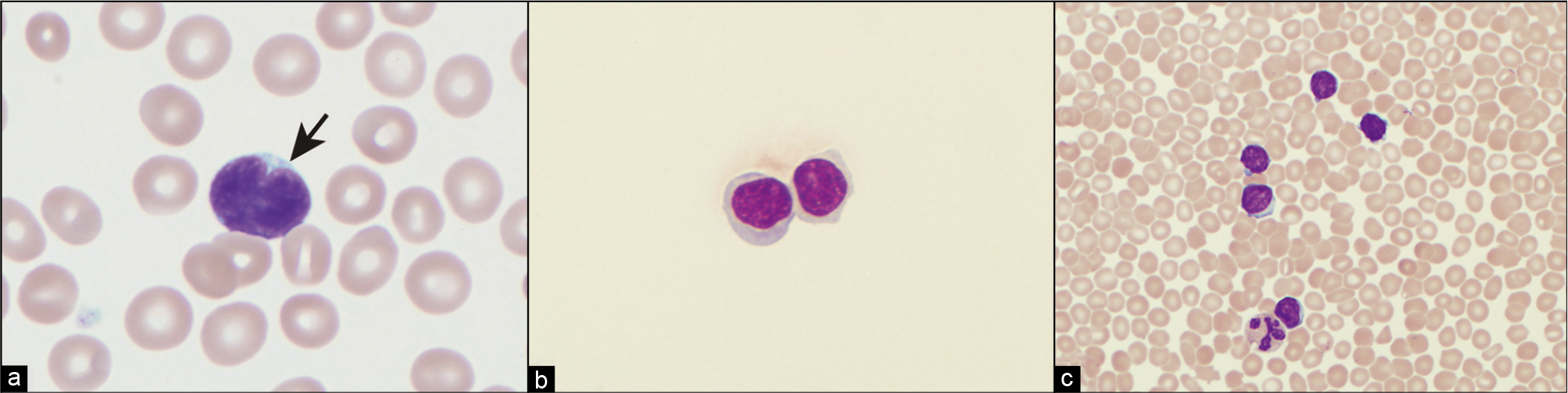
- (a) Follicular lymphoma lymphocyte with characteristic cleaved or notched nucleus (arrow). (b) CLL cells in pleural fluid Cytospin preparation. (c) Bloody pleural fluid in a patient with CLL. Contamination with peripheral blood must be considered before diagnosing a patient as having pleural fluid involvement with CLL.
![Burkitt lymphoma, ascitic fluid; monomorphous population of medium-sized cells with prominent cytoplasmic vacuolization. Note the mitoses in C (arrows). [DQ stain].](/content/105/2022/19/1/img/Cytojournal-19-17-g002.png)
- Burkitt lymphoma, ascitic fluid; monomorphous population of medium-sized cells with prominent cytoplasmic vacuolization. Note the mitoses in C (arrows). [DQ stain].
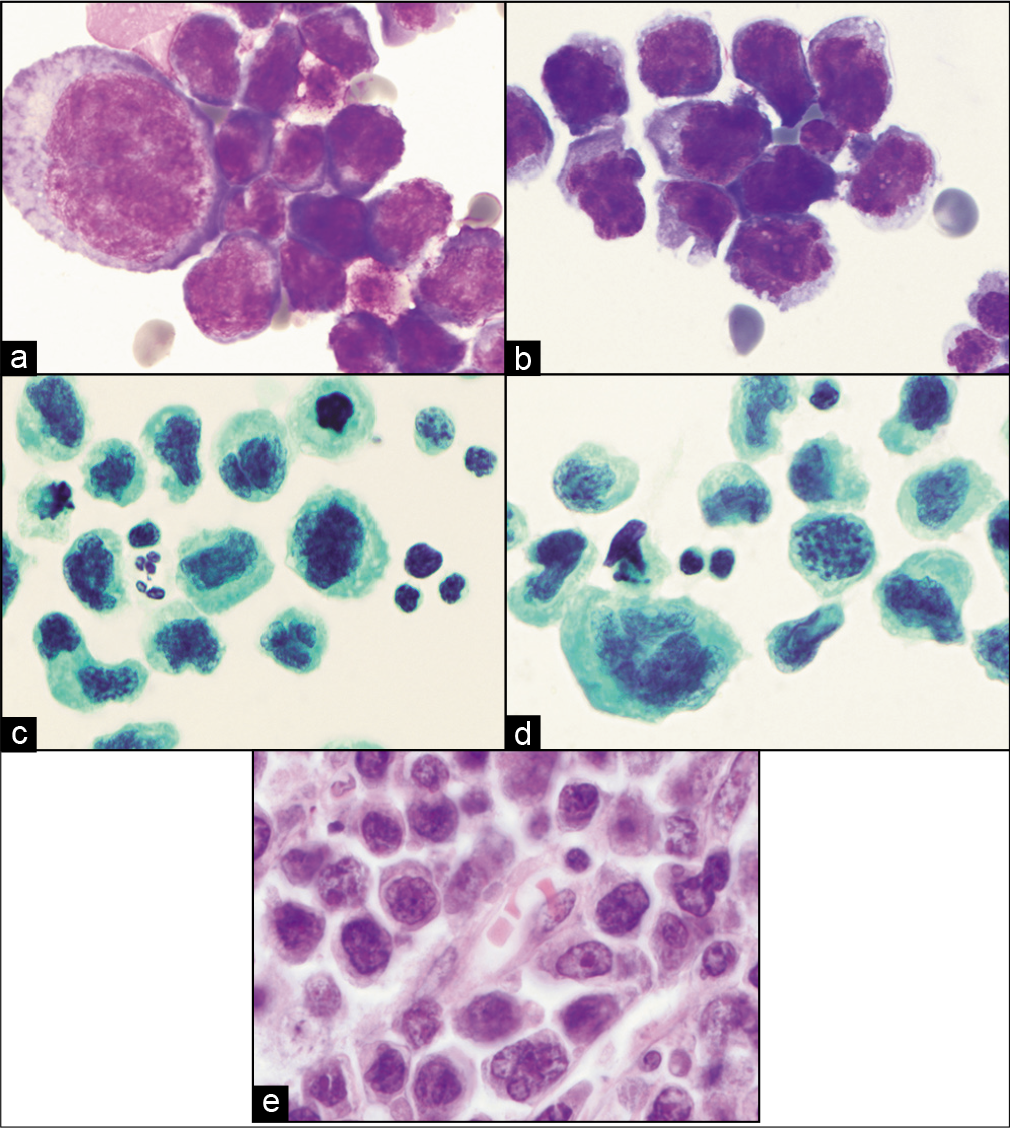
- (a,b) DQ-stained cytology smears from ascitic fluid showing large atypical lymphoid cells later proved to be diffuse large B-cell lymphoma. (c,d) PAP-stained cytology smears showing same cells as in a and b. (e) Histology section from same patient with malignant effusion showing primary colonic diffuse large B-cell lymphoma.
| Stain | CHL (RS cells) | CLL/SLL | FL | MCL | MZL | LPL | ALCL | DLBCL | NLPHL | NKL | PCM | PEL | BL | B-ALL/LBL | T-ALL/LBL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3 | -/rare + | - | - | - | - | - | - (75%) | - | - | +*/- | - | - rare + |
- | - | ± |
| CD4 | -/rare + | - | - | - | - | - | + | - rare + |
- | ± | - | - | - | - | ±** |
| CD5 | -/rare + | + | - | + rare - |
- | - | - | + (10%) | - | - | - | - | - | - | ± |
| CD8 | -/rare + | - | - | - | - | - | - | - | - | ± | - | - | - | - | ±** |
| CD10 | - | - | + (60-80%) | - | - | - | - | + (50%) | - | - | ± | ± | + | ± | ± |
| CD15 | + (80%) | - | - | - | - | - | - rare + |
- | - | - | - | - | - | ± | - |
| CD20 | - (80%) | + | + | + | + | + | - | + rare - |
+ | - | - | - | + | ± | - |
| CD23 | - | + rare - |
± | - rare + |
± | - | - | - | - | - | - | - | - | - | - |
| CD30*** | + | - | - | - | - | - | + | ± | - | - | - | + | - | - | - |
| CD43 | - | + | - (90%) | + | ± | ± | ± | ± | - | + | + (50%) | ± | + | + (75%) | + |
| CD45 | - | + | + | + | + | + | ± | + | + | + | - | ± | + | weak/- | weak/- |
| CD56 | - | - | - | - | - | - | - | - | - | + | ± | ? | - | - | - |
| CD68 | - | - | - | - | - | - | ± | - | - | - | - | - | - | - | - |
| CD79a | - rare + |
+ | + | + | + | + | - | ± | + | - | + | - | + | + | - |
| CD138 | - | - | - | - | ± | ± | -/? | ± | - | -/? | + | + | - | ± | ± |
| ALK | - | - | - | - | - | - | + (85%) | - rare + |
- | - | - | - | - | - | - |
| BCL-1 (cyclin D1) |
- | - | - | + | - | - | - | - | - | - | ± | - | - | - | - |
| BCL-2 | ? | + | +**** | + | ± | + | ? | + (25–50%) |
? | ? | + | ? | - | ± | ± |
| EBV | ± | - | - | - | - | - | - | ± | - | + | - | ± | + (40%) | - | - |
| EMA | - (95%) | - | - | - | - | - | ± | - rare + |
± | - | - | - | - | - | - |
| HHV-8 | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - |
| PAX5***** (BSAP) |
+ (90%) | + | + | + | + | + | - | + | + | - | - | + | + | + | - |
| TdT | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + |
HL/RS = classical Hodgkin lymphoma / Reed–Sternberg cells, CLL/SLL = chronic lymphocytic leukemia/small lymphocytic lymphoma, FL = follicular lymphoma, MCL = mantle cell lymphoma, MZL = marginal zone lymphoma, LPL = lymphoplasmacytic lymphoma, ALCL = anaplastic large cell lymphoma, DLBCL = diffuse large B-cell lymphoma, NLPHL = nodular lymphocyte predominant Hodgkin lymphoma, NKL = natural killer lymphoma, PCM = plasma cell myeloma (multiple myeloma), BL = Burkitt lymphoma, ALL/LBL = precursor T- or B- lymphoblastic leukemia/lymphoma
In contrast to adults, the most common causes of malignant effusions in children are hematopoietic neoplasms. A study of 508 samples from patients under 18 years of age with serous effusions showed that pleural fluids represented the largest group, constituting 61.4% of the fluids, while ascitic fluid and pericardial fluid represented 25.6% and 7.5%, respectively. Of these 508 cases, 226 (44%) had a documented neoplasm. A positive cytology for neoplastic cells was found in 47.5%, 23.1%, and 42.9% from pleural, ascitic, and pericardial fluids, respectively. The most common tumor types associated with a positive cytology in children were lymphoma/leukemia (52%), neuroblastoma (14%), Wilms’ tumor (9%), germ cell neoplasms (8%), bone and soft tissue sarcomas (7%), epithelial neoplasms (5%), and Ewing’s sarcoma (2%). For each site with a positive cytology, the patients had lymphoma/leukemia in 57.6% of pleural fluids, 33.3% of ascitic fluids, and 100% of pericardial fluids. However, it is rare for a child to present with a malignant effusion without first having a tissue diagnosis.[13]
Pleural fluid
Pleural fluid in normal, healthy, non-smoking adults typically consists of macrophages (median 75%), lymphocytes (median 23%), and mesothelial cells (median 1%). Neutrophils (median 0%) and eosinophils (median 0%) are only rarely present. This differential cell count appears to remain constant in patients aged from 17 to 54 years. The total pleural fluid volume in normal adults averages 0.26 ± 0.1 mL per kilogram body mass. Non-smokers have a median total white blood cell (WBC) count of 91 × 103/mL. In smokers, an increase in total WBC count (median 147 × 103/mL) and neutrophils (median 1%) can be seen.[14,15] Normal pleural fluid cytology in children has not been studied to the same extent as it has been in adults but generally shows no marked variation from non-smoker health young adults.
Peritoneal fluid
Many of the hematologic malignancies presenting as peritoneal effusions can also manifest as pleural or even pericardial effusions. Ascites has numerous causes, and often a clinical history will provide clues to the etiology. The most common causes include hepatic cirrhosis, congestive heart failure, renal disease, neoplasms, and infection. However, one should not forget that patients with cirrhosis or congestive heart failure can also have peritoneal involvement with cancer and infectious agents, and thus a careful examination of the effusion fluid is required to rule out superimposed disease processes that may be contributing to the ascites. Evaluation of the fluid requires gross examination, protein estimation, total cell count, differential cell count, Gram stain, acid-fast stain, culture, and cytologic and cell-block examination.[6-8]
Pericardial fluid
The composition of ‘normal’ pericardial fluid has been determined in patients undergoing elective heart surgery for coronary or valvular disease. Patients with prior (within 3 months) myocardial infarction, any known pericardial disease, systemic or autoimmune disease affecting pericardium, or use of medications associated with pericarditis were excluded from the study.[9] These adult patients had a normal total serum WBC count with a normal leukocyte differential. The mean total pericardial WBC count was 1400/μL with a mean of 53% lymphocytes, 31% neutrophils, 12% monocytes, 1.7% eosinophils, and 1.2% basophils. Thus, a diagnosis of ‘lymphocytosis’ in pericardial effusions should be rendered cautiously.[16]
Malignant pericardial effusions in adults are most commonly secondary to lung and breast carcinoma, but metastatic melanoma and lymphoma/leukemia are also common.[11,17] Most patients will have a clinical history of malignancy, but rarely pericardial effusion may be the initial presentation of the disease. Primary cardiac lymphoma and leukemia are very rare and cardiac involvement of systemic lymphoma has been reported in up to 20% of cases.[11,18] Immunocytochemical stains for cytokeratin, S-100 protein, HMB45, and CD45 should be performed when suspicion for a non-epithelial malignant effusion exists.[17] Once a lymphomatous/leukemic effusion is favored by this panel, evaluation of the morphologic and immunophenotypic features described in the following sections should be used for diagnosis and further classification.
As mentioned previously, pericardial effusions in children are rare. In a large study of serous fluid cytology specimens over a 40-year period, only 7.5% of the pediatric specimens were from pericardial fluid. The only positive malignant cytology in pericardial fluids in these patients was due to Hodgkin lymphoma.[13]
Fifteen percent of pericardial effusions were malignant. Hypothyroidism (9%), rheumatoid arthritis (7%), Coxiella burnetii (5%), enteroviruses (4%) and systemic lupus erythematosus (3%) were some other causes; 48% of pericardial effusions did not have an identifiable cause and remained ‘idiopathic.’[19]
LYMPHOCYTIC EFFUSIONS
• A lymphocytic pleural effusion is defined as a lymphocyte differential cell count exceeding 50%.[20] The lymphocytic effusions may be either transudative or exudative and reactive or neoplastic. The variation in type and etiology of the effusion depends on the patient population.
Lymphomatous and leukemic pleural effusions are classically exudative. Cases of transudative effusions are seen in patients who have concomitant diseases such as renal failure or heart failure.[21] Non-Hodgkin lymphoma accounts for approximately 10% of all malignant pleural effusions. Patients with non-Hodgkin lymphoma may develop a pleural effusion in 10–20% of cases[20,22] (positive cytology in up to 60-90%).[22] The reason for negative cytology may be related to: (1) a potentially reactive effusion secondary to obstruction of lymphatic drainage or (2) the patients may have concurrent unrelated disease causing an effusion.[22] Patients with Hodgkin lymphoma may develop benign pleural effusions in up to 20% of cases due to obstruction of mediastinal or pulmonary lymphatic drainage.[23]
Furthermore, non-Hodgkin lymphoma is a common cause of chylothorax, which is an accumulation of lymphatic fluid in the pleural space and is associated with increased pleural fluid triglyceride levels (greater than 110 mg/dL is highly suggestive of a chylous effusion).[24] In over 50% of patients with a malignant effusion associated with a chylothorax, the malignant tumor is a non-Hodgkin lymphoma.[11]
Reactive lymphocytic effusions
Congestive heart failure
In regions with low rates of tuberculosis and/or with a high incidence of heart disease (e.g. the United States), congestive heart failure is the most common cause of lymphocytic effusions in adults. It results in a transudative lymphocytic effusion. Exudative effusions are rare.[25]
Pulmonary embolism
The differential WBC count in patients with pulmonary embolism is more often predominated by neutrophils (60% of patients), but in 40% of patients, lymphocytes predominate. Greater than 10% eosinophils can be seen in up to 18% of effusions secondary to pulmonary embolism. The effusion characteristically is an exudate and frequently shows increased mesothelial cells and red blood cells (RBCs).[26]
Syphilitic pleuritis
A single rare case of syphilitic pleuritis diagnosed by cytopathology reported involvement of the lung and pleura by Treponema pallidum, which resulted in a pleural effusion. The 68-year-old patient described in this report had been diagnosed previously with syphilis.[27] Cytologic examination of the pleural fluid by alcohol-fixed PAP and air-dried Giemsa-stained smears showed many lymphocytes and smaller numbers of plasma cells. Aggregates of histiocytes with foamy cytoplasm were also present. In the Giemsa-stained preparations, rare purple-stained spirochetes could be seen free in the background and in the cytoplasm of histiocytes. In the later stages of syphilis, epithelioid granulomas may appear and the distinction from tuberculosis and sarcoidosis may be difficult. Dark-field illumination or immunofluorescent microscopy are superior methods to Giemsa staining in detecting spirochetes. In suspected cases, real-time PCR for Treponema pallidum is more reliable in confirming the diagnosis of primary syphilis.
Viral pericarditis
Viral pericarditis is most commonly caused by Coxsackie B virus. Coxsackie virus, which belongs to the genus of enteroviruses, may cause a rapidly fatal myopericarditis. In some cases, the lymphocytes in the effusion fluid may be highly reactive and appear with atypical features.[28] The differential diagnosis in such cases includes cardiac lymphoma, and ancillary studies should be performed to rule out a involvement by lymphoma and leukemia cells.
Tuberculosis
In regions with high tuberculosis rates, the most common etiology of lymphocytic pleural effusions is pulmonary infection with Mycobacterium tuberculosis.[12,29-31] Tuberculosis is associated with an exudative effusion containing numerous (>50%) lymphocytes and usually very few mesothelial cells. • The small lymphocytes may show reactive changes, numerous and difficult to distinguish from lymphoma or leukemia cells. In tuberculosis, the lymphocytes are virtually all of T cells type. Multinucleated giant cells are usually not seen in pleural fluid cytology but may be seen in 60–80% of pleural biopsies.[11,32] The lymphocytes in tuberculous effusions can form lymphoid groups or clusters, presumably because of fibrin trapping.[1] In some patients, pleural fluid may contain greater than 10% eosinophils (see section on eosinophilic effusions) and increased numbers of mesothelial cells.[30]
The success rate in diagnosing tuberculous peritonitis is disproportionately low. This may in part be due to a toolow index of suspicion in patients with ascites. The gross appearance of the fluid is highly variable from clear to turbid to hemorrhagic. In 7% of cases, the RBC count may exceed 10,000/μL. In 70% of cases, the leukocyte count is greater than 1000 WBCs per μL. Reactive T cells constitute greater than 70% of nucleated cells. As with tuberculous pleuritis, the best method for diagnosis is biopsy. Histologic examination usually shows granulomas and acid-fast bacilli.
Other causes of reactive lymphocytic effusions
Other causes of non-neoplastic lymphocytic effusions include infarction, drug reactions, and various types of pneumonia.[33]
Neoplastic lymphocytic effusions [Table 2]
Chronic lymphocytic leukemia/small lymphocytic lymphoma CLL/SLL
CLL/SLL is the most common leukemia in adults over 50 years old.[34,35] CLL and SLL represent two ends of the spectrum of a single disease distinguished only by the main site of neoplastic involvement. CLL by definition involves the peripheral blood and bone marrow at the time of diagnosis. Approximately 3.5% of CLL/SLL patients will eventually develop a high-grade lymphoma (Richter syndrome), which is most often a diffuse large B-cell lymphoma. Sheets of immunoblasts suggest transformation to a diffuse large B-cell lymphoma. CLL/SLL patients may also develop Hodgkin lymphoma in 0.5% of cases.[34]
Pulmonary complications are a common cause of morbidity and mortality in CLL/SLL patients. The most common pulmonary illnesses are pneumonia (75%), followed by malignant pleural effusion due to CLL/SLL (5%). • In many cases with clinically suspected serous cavity involvement, if flow cytometry is not performed, the distinction from reactive lymphocytes may be difficult by cytology alone.[36] Thus, the true incidence of malignant effusion secondary to CLL/SLL may be underestimated.
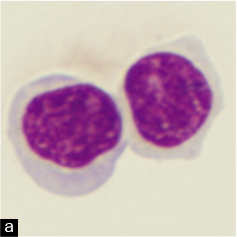
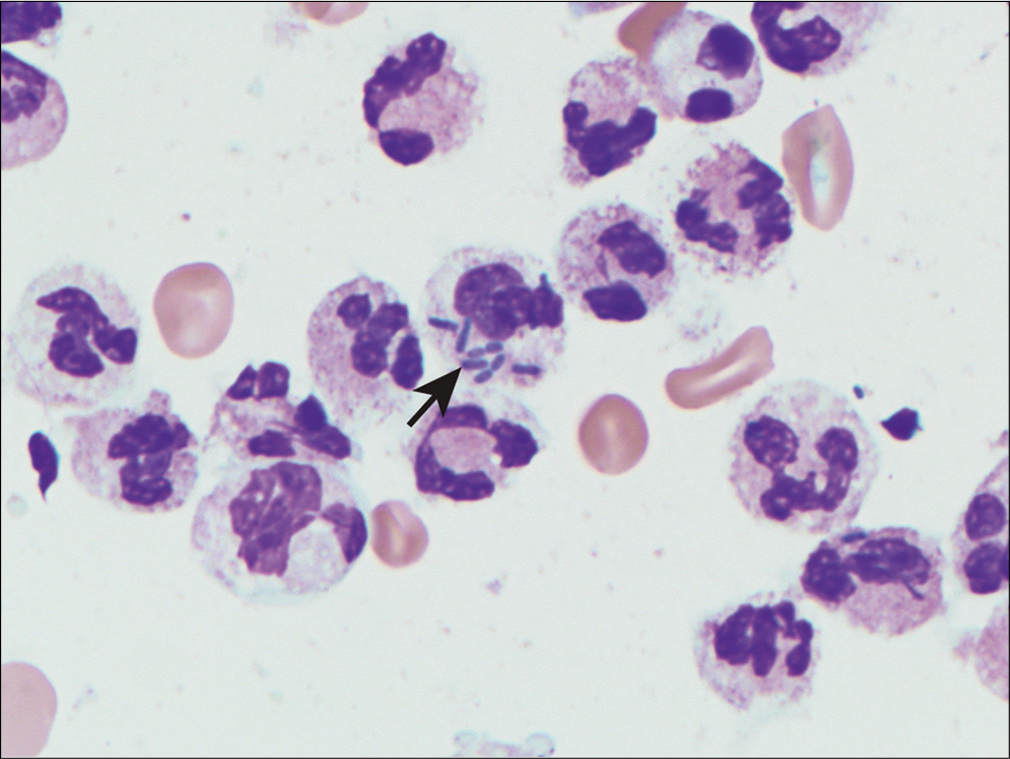
The classically described cytomorphology of CLL/SLL [see Table 1] is that of a neoplasm composed of mostly monomorphic, small, round lymphocytes with clumped chromatin, scant cytoplasm, and indistinct or absent nucleoli [Figure 1b,c]. The chromatin pattern has been described as ‘soccer-ball-like.’ Compare the CLL/SLL lymphocyte with the cleaved centrocyte of a germinal center or in follicular lymphoma [Figure 1a]. In tissue, CLL/SLL, however, is composed of varying numbers of prolymphocytes, paraimmunoblasts, and plasmacytoid lymphocytes that differ from the classically described small round lymphocyte. In tissue sections, these prolymphocytes and paraimmunoblasts collect to form ‘pseudo-follicles’ or ‘proliferation centers’ that can readily be seen as lighter areas on low-power magnification. Prolymphocytes are medium sized (approximately twice the size of a small lymphocyte), have a small but distinct nucleolus, and have an increased amount of cytoplasm. Paraimmunoblasts are medium to large cells (2–3 times a small lymphocyte) with round to oval nuclei, multiple peripheral nucleoli or a single large prominent central nucleolus, and slightly basophilic cytoplasm on Wright–Giemsa stain.[34,37]
• The challenge in diagnosing a malignant effusion due to CLL/SLL stems from (1) distinguishing it from a reactive lymphocytosis and (2) determining whether there is contamination with peripheral blood lymphocytes secondary to a traumatic/bloody thoracentesis. The lymphocytes of CLL/SLL can be morphologically indistinguishable from reactive lymphocytes. This is because, as described above, the lymphocytes in CLL/SLL can be heterogeneous in the same way as reactive lymphocytes. A monomorphic population of small round lymphocytes, though, is suggestive of involvement by CLL/SLL.
Further support for diagnosis of CLL/SLL can be attained by making a cell-block from extra serous fluid and performing immunohistochemical stains.[7] • Usually, reactive lymphocytes are predominantly CD3 positive T cells while a malignant pleural effusion due to CLL/SLL usually shows predominantly CD20 positive B cells. Aberrant expression of CD5 (the T-cell marker) in the B cells with co-expression of CD23 without cyclin-D1 will help confirmation of involvement by CLL/SLL [see Table 1]. • Furthermore, flow cytometry studies are extremely helpful and also much more sensitive at identifying a small population of monoclonal B cells that may not be evident by cytology or immunohistochemistry.
• Caution should be exercised before making a diagnosis of a malignant CLL/SLL effusion until the possibility of a traumatic thoracentesis has been ruled out. This can be done by examining the fluid for significant numbers of RBCs. Blood containing lymphoma cells can result in a false-positive diagnosis.[32] It is also possible that the leukemic/lymphomatous infiltrate may directly infiltrate the pleura without producing a frankly malignant effusion. In this case, one will see reactive T cells in the serous fluid and a neoplastic B-cell infiltrate in the biopsy.[38,39]
Other ‘small’ B-cell lymphomas: follicular, mantle cell, marginal zone lymphoma, lymphoplasmacytic lymphoma
The previous discussion of malignant effusion diagnoses in CLL/SLL also applies to other small B-cell lymphomas such as follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma (including mucosa-associated lymphoid tissue lymphoma—MALT lymphoma). • That is to say, reactive versus neoplastic lymphoid proliferations of small lymphocytes in these lymphomas may be difficult, if not impossible, to separate by cytologic examination alone. The diagnostic accuracy for all lymphomas, even by FNA, varies from 64 to 91%, but for follicular lymphomas the diagnostic accuracy varies from 37 to 69%[2] • Immunohistochemistry, flow cytometry, and molecular studies are useful and may be critical ancillary tools when examining small lymphocytic neoplasms.
Follicular lymphoma
Follicular lymphoma is the second most common lymphoma in the United States (35% of lymphoma) and worldwide (22% of lymphoma).[34,35] It is twice as common in whites as in blacks. Only 9% of follicular lymphomas show extranodal involvement.[40]
Cytomorphologically, the characteristic lymphocyte of follicular lymphoma is the small-cleaved lymphocyte, also known as a centrocyte, which has a small indented nucleus, indistinct to absent nucleolus, and scant cytoplasm [see Figure 1a, Table 1]. The characteristic feature of irregular nuclei in follicular lymphoma has been described as nuclear notches, nuclear projections, and nuclear clefts. In some cases, the nuclear irregularities may be subtle.[37] However, the neoplasm is also composed of varying numbers of centroblasts, which are large cells with one to three small, peripherally located nucleoli attached to the inner part of the nuclear membrane.
• As the grade of the lymphoma increases, the number of centroblasts also increases. Therefore, a heterogeneous population does not imply benignity. However, in many cases, centrocytes predominate (grade 1-2 follicular lymphoma), and thus give a monomorphic appearance. With grade 3 follicular lymphoma, centroblasts may predominate to the point of making the distinction from diffuse large B-cell lymphoma difficult. In most patients, however, grade 3 follicular lymphoma and diffuse large B-cell lymphoma are treated the same.[41]
• Immunophenotypically, the lymphocytes of follicular lymphoma are positive for CD20, BCL-2, BCL6 and CD10, while negative for CD5.[34] The immunophenotypic findings are critical in confirming the diagnosis; however, they will not help in distinguishing a CD10-positive diffuse large B-cell lymphoma from a grade 3 follicular lymphoma [see Table 1]. In addition, grade 3 follicular B cell lymphoma can be BCL-2 negative thus making it difficult to differentiate from Burkitt lymphoma.
Extranodal marginal zone lymphoma/mucosa-associated lymphoid tissue (MALT) lymphoma.
Extranodal marginal zone lymphoma represents 7–8% of non-Hodgkin lymphomas (NHLs).[34,35] MALT lymphoma is common in the stomach and is associated with Helicobacter pylori infection. It also frequently involves the lung (14% of MALT lymphoma cases) and salivary gland, and often associated with autoimmune diseases.
Cytomorphologically, the characteristic cell of marginal zone lymphoma is small to medium in size, has an irregular nucleus with inconspicuous nucleolus, and has abundant pale cytoplasm, and monocytoid B cell morphology. The abundant pale to clear cytoplasm may give the cell a monocytic appearance, and thus the term monocytoid has been traditionally used to describe these cell. DQ-stained slides are often best at enhancing subtle differences in cell size and quantity of cytoplasm. However, monocytoid cells may be in the minority, as cells of marginal zone lymphoma can resemble virtually any lymphocyte subset, including centrocytes, centroblasts, and may have increased immunoblasts. In addition, they may undergo plasmacytic differentiation. Thus, MALT lymphoma is classically described as being composed of a heterogeneous population of B cells, including centrocytelike and monocytoid-like cells, and scattered immunoblasts and centroblast-like cells. There may also be a background population of reactive T-cells. • The heterogeneous lymphoid population in MALT lymphoma, in contrast to the classic monomorphic appearance of lymphoma cytology, can be misleading and result in a diagnosis of a reactive process.
• Immunophenotypically, the diagnosis of marginal zone lymphoma/MALT lymphoma is one of exclusion since currently there is no specific immunophenotype. Marginal zone lymphoma/MALT lymphoma is positive for CD20 and negative for most other markers (CD10, CD5, and BCL-1/ cyclin D1, CD23 and). The neoplastic cells may express CD23 and the T cell/myeloid marker CD43), a feature supportive of a neoplastic process. This finding is particularly useful when flow cytometry or molecular studies are not available to establish the clonality. Occasionally, they may express CD11c which of low significant diagnostic value.[34,42]
Mantle cell lymphoma
Mantle cell lymphoma represents 3–10% of non-Hodgkin lymphomas. Despite its histologic resemblance to low-grade small cell lymphomas, it is considered an intermediate-grade lymphoma with a poor prognosis (median survival 3–5 years). The most common extranodal sites include bone marrow (>50%), gastrointestinal tract (30%), peripheral blood (25%), spleen, lung, pleura, and cerebrospinal fluid (9%).[34,43,44]
Cytomorphologically, in body fluid and in blood, the cells of mantle cell lymphoma typically resemble prolymphocytes of B prolymphocytic leukemia.[37] In some cases, the small cleaved lymphocytes may closely resemble a follicular lymphoma centrocyte and thus a diagnosis of mantle cell lymphoma can be difficult, if not impossible, by cytologic examination alone.[37] Mantle cell lymphoma is characterized by a monotonous population of small lymphocytes without admixed centroblasts or paraimmunoblasts. However, larger cells with immature lymphoblastic morphology (fine chromatin, nucleoli, and little cytoplasm) or larger, more pleomorphic cells with irregular nuclei and prominent nucleoli may appear in the so-called ‘blastoid’ variant of mantle cell lymphoma. The blastoid variant of mantle cell lymphoma is associated with an aggressive course.
• Immunophenotypically, the lymphocytes of mantle cell lymphoma can be shown to express nuclear BCL-1 (cyclin-D1), CD20, and the T-cell-associated markers CD5 and CD43. Cases of CD5 negative mantle cell lymphoma have been described and thus when mantle cell lymphoma is suspected, one should order immunohistochemistry for cyclin-D1. The neoplastic cells are characteristically CD10 negative, a feature distinguishing mantle cell lymphoma from follicular lymphoma. These cells are usually CD23 negative, in contrast to CLL/SLL. In cases with inconclusive immunophenotypic findings, fluorescent in-situ hybridization (FISH) for the t(11:14) translocation is extremely helpful to make the diagnosis. A subset of mantle cell lymphoma is cyclin-D1 negative and t(11;14) negative. For such cases SOX11 immunostain is positive and should be utilized.
Lymphoplasmacytic lymphoma/Waldenström macroglobulinemia
Lymphoplasmacytic lymphoma is an exceedingly rare lymphoma that is largely a diagnosis of exclusion when the criteria for other low-grade, plasmacytoid, small B-cell lymphomas cannot be met. Lymphoplasmacytic lymphoma is mostly known for its association with Waldenström macroglobulinemia. However, other leukemias and lymphomas, such as CLL/SLL and marginal zone lymphoma, can be associated with a serum monoclonal IgM protein as well and may have plasmacytic differentiation.
The cells of lymphoplasmacytic lymphoma consist of a mixture of small and larger lymphocytes with plasmacytoid features (basophilic cytoplasm, perinuclear clearing, eccentric nucleus). Dutcher bodies (intranuclear inclusions) and Russell bodies (intracytoplasmic inclusions) may be seen as a manifestation of accumulated immunoglobulins. Cytoplasmic light chain restriction can often be demonstrated by immunocytohistochemistry. Kappa restriction is more common than lambda.[37]
LYMPHOPLASMACYTIC LYMPHOMA/WALDENSTRÖM MACROGLOBULINEMIA, defined by the 2017 World Health Organization (WHO), is a neoplasm involving bone marrow in which the neoplastic cells are positive for surface IgM and CD20 and negative for CD5, CD10, and CD23. By morphology alone, it may be difficult or impossible to differentiate lymphoplasmacytic lymphoma from marginal zone lymphoma without knowing bone marrow status or serum protein electrophoresis results. Lymphoplasmacytic lymphoma and marginal zone lymphoma appear closely related and they may reflect a spectrum of one disease.[45,46]
Burkitt lymphoma
Burkitt lymphoma is a mature B-cell malignancy that occurs most commonly in children (sporadic variant), immunodeficient patients (primarily human immunodeficiency virus (HIV), and in parts of Africa (endemic variant). Majority cases of Burkitt lymphoma have a translocation of the MYC gene on chromosome 8. The most common translocation is t(8;14) but less commonly t(2;8) and t(8;22) may occur. Burkitt lymphoma also has interesting historical and etiologic association with Epstein–Barr virus (EBV). EBV was first discovered in Burkitt lymphoma cells sent to Epstein’s laboratory from Africa. These cells came from the endemic variant of Burkitt lymphoma, which has been shown to contain EBV DNA in 95% of cases. EBV is also seen in 25–40% of immunodeficiency-related cases and in 20% of sporadic cases of Burkitt lymphoma.[34,47]
The sporadic variant of Burkitt lymphoma occurs mostly in children and young adults. Sporadic Burkitt lymphoma represents 30–50% of childhood lymphomas. The majority of patients with sporadic Burkitt lymphoma (80–91% of patients) present with abdominal masses, which represent involvement of peritoneum, omentum, bowel mesentery, or bowel wall. The masses may continue to grow and involve breasts, ovaries, testes, kidney, liver, adrenals, and spleen. [34,47] • Extensive involvement of the peritoneum may result in ascites. Ascites occurs in up to 58% of sporadic Burkitt lymphoma cases in the USA.[47,48]
Spread to pleura and pericardium may similarly give rise to serous effusions at those locations. Pleural and peritoneal effusions are seen more commonly than pericardial effusions. Ascites occurs in up to 58–90% of sporadic Burkitt lymphoma cases in the USA, while pleural effusions occur in 19–67%.[47,48]
The immunodeficiency-related variant of Burkitt lymphoma is primarily associated with HIV infection. In fact, Burkitt lymphoma may be the initial manifestation of acquired immune deficiency syndrome (AIDS) in some HIV-infected individuals.[34] Burkitt lymphoma is more likely to occur early in AIDS, while diffuse large B-cell lymphoma occurs later in the illness. EBV is identified in 25–40% of Burkitt lymphoma cases in HIV-associated cases. Diffuse large B-cell lymphoma has a stronger association with EBV in AIDS patients than does Burkitt lymphoma. EBV is identified in 60% of all HIV related lymphomas. Burkitt lymphoma represents 30% of all HIV-associated lymphomas.[34,47]
• The diagnosis of Burkitt lymphoma can be an oncologic emergency, owing to its near 100% proliferation rate. The tumor cells have a doubling time of only 24 hours. This high growth rate may lead to ‘tumor lysis syndrome’ after initiation of therapy, which may cause renal failure and sudden hyperkalemia. Serous effusion cytology is the ideal specimen for this lymphoma in which a rapid diagnosis is required. Burkitt lymphoma doesn’t commonly cause pleural and peritoneal effusions for which cytologic examination is possible.[48]
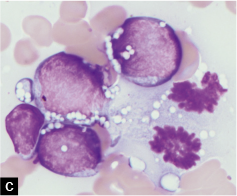
Characteristically, Burkitt lymphoma is composed of strikingly monomorphic, non-cohesive, non-cleaved, medium-sized cells with regular nuclei, and prominent cytoplasmic vacuoles [see Table 1]. • The cytoplasmic vacuoles are most readily apparent on air-dried Wright– Giemsa or DQ-stained smears [Figure 2]. While air-dried smears demonstrate excellent cytoplasmic detail, the monomorphism of the cells is less apparent with these preparations, which accentuate subtle differences in cell size and amount of cytoplasm, making them appear more pleomorphic than in alcohol-fixed smears or tissue sections. The characteristic cytoplasmic vacuoles in Burkitt lymphoma cells represent cytoplasmic lipid that can be stained with lipid stains such as Oil Red O. The chromatin pattern is clumped and many cells contain multiple (often 2–5) easily identifiable nucleoli. The thin rim of surrounding cytoplasm is deeply basophilic owing to the high RNA content. Mitotic figures may be readily identified [see Figure 2c]. One may also see ‘tingible-body macrophages’ which have phagocytosed cellular debris from the rapidly dividing cells. These tingible-body macrophages impart the ‘starry-sky’ appearance seen in tissue sections and can be seen in any rapidly proliferating lymphoma and leukemia.
• The gold standard for the diagnosis of Burkitt lymphoma is the MYC gene translocation, which most commonly involves t(8;14), in the setting of a high-grade lymphoma with Burkitt or Burkitt-like morphology (i.e. relatively monomorphic cells with nuclei smaller than a macrophage nucleus) along with the appropriate immunophentotype. Many large reference laboratories can test for the MYC translocation using FISH on paraffin-embedded formalin-fixed tissue.
The cells of Burkitt lymphoma are positive for CD19, CD20, CD22, and usually CD43. They also express the germinal-center-associated antigens CD10 and BCL-6. The cells are negative for TdT, CD5, CD23, and BCL-2. The cells also express surface IgM and light chain detectable by flow cytometry. Nearly 100% of the cells are positive for the Ki-67 (MIB-1) proliferation marker. It is generally accepted that a high MIB-1 (KI67) approaching 100%, expression of CD20 and CD10, and lack of BCL-2 and TdT expression, along with the appropriate cytomorphology, are sufficient for a presumptive diagnosis of Burkitt lymphoma. Those Burkitt lymphomas associated with EBV may show positive staining with EBV-LMP or EBER in-situ hybridization. CD21 is also expressed more commonly in cases associated with EBV. In tissue, infiltrating background T cells are less common than in diffuse large B-cell lymphomas, but in effusion cytology background reactive T cells may be abundant.
Diffuse large B-cell lymphoma [Figure 3]
Diffuse large B-cell lymphoma is a heterogeneous group of high-grade, aggressive B-cell lymphomas that accounts for 40% of non-Hodgkin lymphomas in United States. It occurs more frequently in men and in whites. Diffuse large B-cell lymphoma may arise either de novo or as a secondary lymphoma resulting from transformation of a low-grade B-cell lymphoma (e.g. CLL/SLL, follicular lymphoma, MALT lymphoma, etc.).
Diffuse large B-cell lymphomas show extranodal involvement in 20–30% of cases. Involvement of the mediastinum with diffuse large B-cell lymphoma may result in a pleural effusion either by compression of regional lymphatics or by direct involvement of pleura. It may cause ascites when the peritoneum, retroperitoneum, abdominal, or pelvic organs are primarily or secondarily involved by lymphoma. Diffuse large B-cell lymphoma is the most common retroperitoneal malignancy.[49] Ovarian diffuse large B-cell lymphoma, the most common gynecologic non-Hodgkin lymphoma, may rarely cause ascites. Most cases of ovarian lymphoma represent secondary involvement by systemic lymphoma; 10% of ovarian non-Hodgkin lymphomas are primary.[50] One must also distinguish the blastoid variant of mantle cell lymphoma from diffuse large B-cell lymphoma. The differential diagnosis of histologically similar non-lymphoid neoplasms includes granulosa cell tumor, dysgerminoma, small cell carcinoma, and myeloid sarcoma. Immunocytohistochemistry and/or flow cytometry is invaluable in making the diagnosis.
Cytomorphologically, diffuse large B-cell lymphoma can assume a wide array of cytologic features [see Figures 3, 5; Table 1]. By definition, most of the cells are large (>2 times a small resting lymphocyte or the size of a macrophage). The cells may assume a plasmacytoid, anaplastic, or immunoblastic morphology, but they more commonly have centroblastic features. Centroblasts have irregular to oval vesicular nuclei with fine chromatin, 2–4 peripherally located nucleoli, and scant cytoplasm. Immunoblasts have a single, large, centrally-located, prominent nucleolus. T-cell-rich/ histiocyte-rich diffuse large B-cell lymphoma may have large neoplastic multilobated cells that resemble Reed–Sternberg (RS) cells in a background of reactive small T lymphocytes.
![HHV-8 positive primary effusion lymphoma (ascitic fluid). The specimen shows monomorphic, dispersed, medium to large cells with moderate amount of basophilic cytoplasm with scant cytoplasmic vacuoles. Nuclei are round to slightly irregular with fine chromatin and variable prominence of nucleoli. [Wright–Giemsa stain.]](/content/105/2022/19/1/img/Cytojournal-19-17-g004.png)
- HHV-8 positive primary effusion lymphoma (ascitic fluid). The specimen shows monomorphic, dispersed, medium to large cells with moderate amount of basophilic cytoplasm with scant cytoplasmic vacuoles. Nuclei are round to slightly irregular with fine chromatin and variable prominence of nucleoli. [Wright–Giemsa stain.]
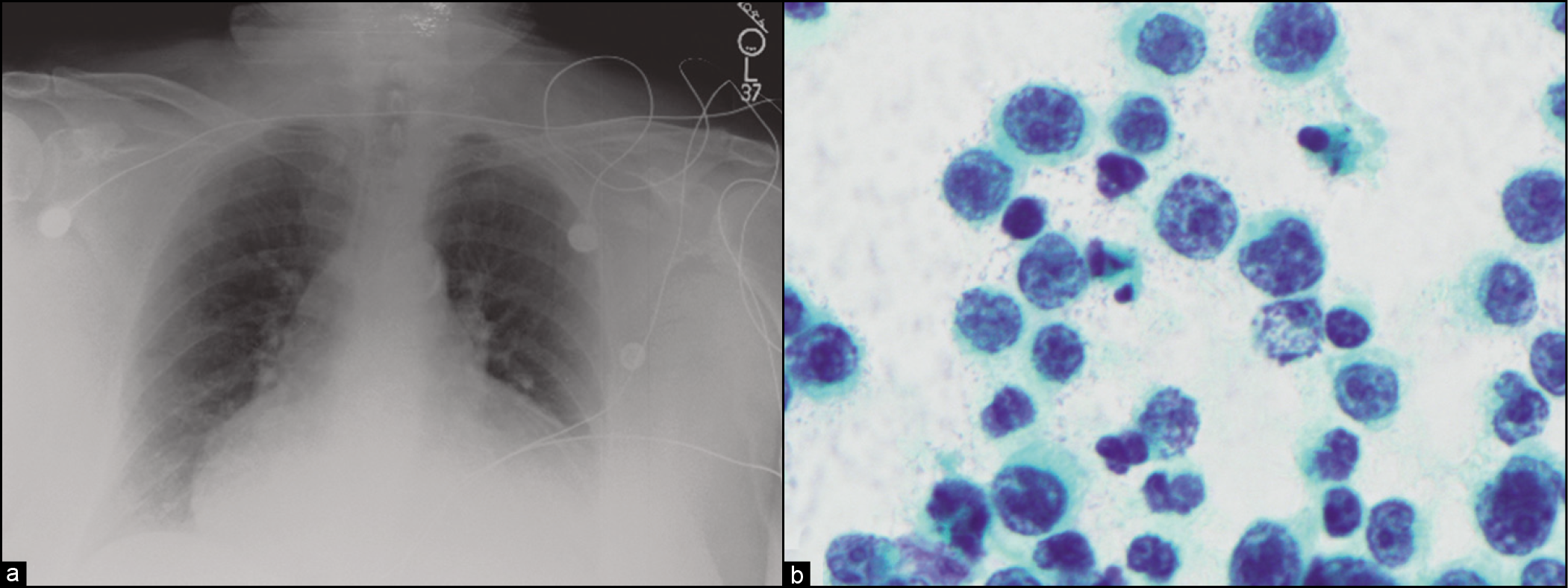
- (a) Chest radiograph showing cardiomegaly in a patient with severe dyspnea and massive malignant pericardial effusion. (b) Pericardial fluid cytology, in patient from (a), presenting with primary cardiac lymphoma and isolated massive pericardial effusion. This case turned out to be diffuse large B-cell lymphoma.
Immunophenotypically, the cells may variably express pan-B markers such as CD20, CD79a, and PAX5. In most cases, the cells are also CD45 positive. Cells with anaplastic morphology often express CD30, but non-anaplastic cells may also occasionally express CD30. EMA is usually negative but may be positive in some cases with anaplastic or multilobated RS-like morphology. Some cases may express CD5 (10%) and CD10 (25–50%). BCL-1/cyclin-D1 is negative in the CD5 positive cases, distinguishing it from the blastoid variant of mantle cell lymphoma. BCL-2 is positive in 25–50% of cases. Plasma-cell-associated markers such as CD138 may be seen in a small minority of cases. Ki-67/MIB-1 staining is usually seen in greater than 40% of cells and may be greater than 90% in some cases. Diffuse large B-cell lymphomas seen in HIV patients are not uncommonly positive for EBV and negative for CD20 and CD45.[34]
High grade B cell lymphoma
This lymphoma is a new entity in the WHO 2017 classification. The cells size are variable from small to medium to large and the diagnosis is exclusively defined by the identification of CMYC and BCL-2 translocation (double hit lymphoma) or CMYC/BCL-2 and BCL-6 translocations (triple hit lymphoma) by FISH studies. The MIB1/KI67 is very high in this group and morphologically it may be indistinguishable from Burkitt lymphoma with cytoplasmic vacuoles. Immunophenotypically, they express B cell markers like diffuse large B cell lymphoma.
Primary effusion lymphoma
Primary effusion lymphoma is defined by the WHO as a subtype of diffuse large B-cell lymphoma which primarily presents as a body cavity effusion. The majority of patients have AIDS or have been HIV positive for several years, but HIV-negative elderly patients are also reported.[51,52] • Primary effusion lymphoma is associated with human herpes virus 8 (HHV-8), the same virus associated with Kaposi sarcoma and multicentric plasma cell variant of Castleman’s disease.[34] In rare cases, it can present and manifest as a solid tumor, in which case it has been referred to as primary effusion lymphoma-like solid lymphoma, HHV-8-associated solid lymphoma, and solid primary effusion lymphoma. In some cases, both EBV and HHV-8 are detected in tumor cells, but in virtually all cases, HHV-8 is detected.[53-55] Immunoglobulin gene rearrangement can sometimes be detected. Cases with both T-cell and B-cell gene rearrangement have been reported (gene infidelity). [54,56] Recently, an HHV-8 positive case with only T-cell receptor gene rearrangement was described in a lymph node with concurrent peritoneal effusion, illustrating so-called solid primary effusion lymphoma and T-cell primary effusion lymphoma.[54] Recent reports of HHV-8 negative pericardial and peritoneal primary effusion lymphomas have raised the question as to whether other viruses such as hepatitis C virus (HCV) may be implicated in the pathogenesis of some cases of primary effusion lymphoma. [57,58] The prognosis of primary effusion lymphoma is generally poor, and death ensues within months.[56]
Pericardial primary effusion lymphoma is the rarest form of primary effusion lymphoma.[52]
The cells of primary effusion lymphoma may appear centroblastic, immunoblastic, lymphoblastic, plasmacytic, or anaplastic. Centroblastic cells have one to three small peripherally located nucleoli and round to oval nuclei [Figure 4]. Immunoblastic cells are medium to large in size (2–3 times a small lymphocyte) with round to oval nuclei, a large prominent central nucleolus, and slightly basophilic cytoplasm on Wright–Giemsa stain. Lymphoblastic cells are medium in size, have little cytoplasm, and often have one or more prominent nucleoli. Anaplastic cells may resemble the ‘hallmark’ cells of anaplastic large cell lymphoma.
Immunophenotypically, the typical primary effusion lymphoma associated with HIV and HHV-8 lacks B-cell markers CD20, CD19, and CD79a, but commonly shows positive immunostaining with CD45, CD30, CD38, and CD138, as well as HHV-8. Aberrant CD3 staining has been reported in rare cases. EBV virus by in-situ hybridization (EBER) can often be detected; EBVLMP is often negative. Surface and cytoplasmic immunoglobulin expression are often absent.[34]
A similar lymphoma, pyothorax-associated lymphoma, is in the differential diagnosis of primary effusion lymphoma and may occasionally be confused with this entity. Pyothoraxassociated lymphoma, however, more closely resembles the typical diffuse large B-cell lymphoma in immunophenotype and epidemiology. It is not typically associated with HIV or HHV-8, but is commonly associated with EBV. It characteristically occurs in the setting of chronic pleural inflammation and may represent the so-called HHV-8 negative primary effusion lymphomas described in the literature. It has also been suggested that hepatitis C virus and EBV may be responsible for some cases of so-called peritoneal HHV-8 negative primary effusion lymphomas or peritoneal pyothorax-associated lymphomas.[53]
Plasma cell myeloma (PCM)
Myelomatous effusions are rare (only 80 reported cases) and usually occur in the late stages of the disease.[59] In a study of 1406 patients at one institution, only 7 patients were found to have developed a malignant myelomatous pleural effusion. Myelomatous ascites is rare. Patients with plasma cell myeloma may develop malignant ascites from direct peritoneal seeding or infiltration from hepatic, splenic or intestinal involvement. In other instances, amyloid deposition in the liver may produce portal hypertension and ascites. One must also keep in mind that amyloidosis of the heart and kidney can also contribute to ascites formation.[60]
Diagnostic difficulty arises when plasma cells appear mature or when they are present in low numbers. Flow cytometry is a helpful and more sensitive method than routine cytologic examination for confirming serous fluid involvement by a clonal plasma cell population.
The malignant plasma cells, in air-dried, DQ-stained smears, often have an inconspicuous or absent perinuclear ‘hof ’ (due to a prominent Golgi apparatus) and basophilic cytoplasm. The nuclei are almost always eccentrically located and may be round/oval or pleomorphic. The chromatin is generally coarse and prominent nucleoli are often present.
• The ‘clock-faced’ chromatin pattern typical of the benign plasma cell may not be present.[61] The presence of nucleoli and nuclear immunoglobulin inclusions (Dutcher bodies) in plasma cells usually indicates a neoplastic process, while cytoplasmic immunoglobulin inclusions (Russell bodies) are commonly seen in both reactive and neoplastic processes.
• Both benign and neoplastic plasma cells are usually CD138 and CD79a positive by immunohistochemistry. However, CD138 may be positive in some lymphomas and even carcinomas. Less frequently, neoplastic plasma cells may aberrantly express CD117 , CD13 and CD33. Plasmacytoid tumor cells from metastatic melanoma, breast cancer, and carcinoid tumor should be considered in the differential diagnosis of a malignant pleural effusion.
Acute lymphoblastic leukemia (ALL)/lymphoblastic lymphoma
ALL and lymphoblastic lymphoma, also known in the WHO classification as lymphoblastic leukemia/lymphoma, represent neoplasms with identical cell origins, but different clinical manifestations. In general, when the lymphoblasts (precursor B or T cells) involve blood or bone marrow, the term ALL is used. In contrast, when the neoplasm manifests in nodal or other extranodal tissues, the disease is termed lymphoblastic lymphoma. For diagnostic purposes, cases with extramedullary disease and fewer than 25% lymphoblasts in the marrow are termed lymphoblastic lymphoma. ALL is the most common malignancy of childhood. It accounts for 80% of childhood acute leukemias and 20% of adult acute leukemias. Approximately 80–85% of ALLs are of the precursor B-cell phenotype. T-ALL constitutes 15% of childhood ALL and 25% of adult ALL. ALL in children generally has a better prognosis than ALL in adults. Lymphoblastic lymphoma is more commonly of the precursor T-cell phenotype, representing 85–90% of lymphoblastic lymphomas. Thus, B-lymphoblastic lymphoma is an uncommon type of lymphoma. Both T and B-lymphoblastic lymphoma occur predominantly in patients under 18 years of age. Approximately 50% of T-lymphoblastic lymphomas present as a mediastinal mass. Mediastinal masses with B-lymphoblastic lymphoma are infrequent.
Lymphoblasts in ALL and lymphoblastic lymphoma vary in appearance from small/intermediate cells with scant cytoplasm to larger cells with moderate amounts of basophilic cytoplasm [see Table 1]. Nucleoli may be indistinct or multiple and prominent. The chromatin pattern is finely dispersed. Mitotic figures are variable in number, but are usually fewer in B-ALL than in T-ALL. Azurophilic cytoplasmic granules may be seen in up to 10% of cases. In some cases, the cells may have eccentric cytoplasmic projections forming so-called ‘hand-mirror cells,’ a finding with no clinical significance. In 1% of patients, hand-mirror cells are a dominant cytologic finding.
• The single most useful marker is terminal deoxynucleotidyl transferase (TdT). While TdT is present in most cases of ALL, it is not specific for the disease. TdT positivity can be seen in up to 20% of acute myelogenous leukemias (AMLs) and is also positive in thymic T-cells. In B-ALL, the lymphoblasts are almost always positive for CD19, PAX5, and CD79a. In contrast, CD20 expression is variable. Precursor T-cell lymphoblasts in T-LBL/ALL variably express CD2 and CD5. CD7 and cytoplasmic CD3 are most often positive, and only CD3 is considered T-cell lineage specific since some AMLs may express CD7 and CD2. Surface CD3 expression may sometimes be absent. The neoplastic cells usually show co-expression or co-deletion of CD4 and CD8. Early T-cell precursor (ETP) lymphoblastic leukemia/lymphoma is added to the 2017 WHO classification of hematolymphoid neoplasm as a high-risk T lymphoblastic leukemia/lymphoma. In addition to CD3 and TdT expression, ETP-ALL/LBL is recognized by being CD1a negative, CD7 positive, CD8 negative and may show CD13 or CD33 expression.
Surface immunoglobulin is characteristically absent in B-lymphoblastic lymphoma/ALL; however, clonal gene rearrangement of the immunoglobulin heavy chain can be shown in most cases. The most common recurring cytogenetic abnormality in childhood ALL is t(12;21)(p13;q22), which correlates with favorable prognosis. The Philadelphia chromosome, t(9:22)(q34;q11), is found in 5% of childhood cases and up to 30% of adult cases and is associated with a poor prognosis. Virtually all patients show T-cell receptor beta-chain gene rearrangement, but this is not specific for T-lymphoblastic lymphoma/ALL but can be used to follow the patient for the presence of minimal residual disease.[34,47]
The differential diagnosis of ALL/lymphoblastic lymphoma usually includes other ‘small round blue cell tumors’ of childhood, as well as other lymphomas (especially Burkitt lymphoma) and leukemias. Other tumors to consider that frequently occur in pleural effusions include neuroblastoma, Wilms’ tumor, germ cell tumor, soft tissue sarcoma, and Ewing’s sarcoma.[13] Rhabdomyosarcoma is one of the small round blue cell tumors frequently misdiagnosed as ALL.[47] Immunophenotyping of these tumors can be very useful for differential diagnosis.
Post-transplant lymphoproliferative disorder (PTLD)
Post-transplant lymphoproliferative disorder (PTLD) is a B-cell (85–95%) or T-cell lymphoid proliferation seen in patients following organ transplantation. PTLD can be divided into polymorphous and monomorphous variants based on morphology. PTLD may spontaneously regress when immunosuppression is reduced, and this happens more often with the polymorphic type. • The polymorphic type is composed of a heterogeneous population of lymphocytes, while the monomorphic type appears more like a typical lymphoma, although either type may show a monoclonal proliferation by flow cytometry or gene rearrangement. Most cases present as nodal or extranodal solid tumors.
Secondary involvement of body cavity by solid-organ PTLD has been more often reported than effusion as a primary manifestation of PTLD. However, approximately 10 cases of primary effusion PTLD have been reported in the literature.[62] In contrast to primary effusion lymphoma, PTLD is not associated with HHV-8 infection. However, both PTLD and primary effusion lymphoma are associated with EBV.
The most common morphology of the B-cell or T-cell monomorphic PTLD is that of medium to large-sized transformed lymphocytes with irregular, convoluted nuclei, prominent nucleoli, and frequent mitoses. The cytologic atypia and monomorphism is usually sufficient to recognize the condition as being neoplastic. These tumors should be classified as B-cell or T-cell lymphomas according to the appropriate WHO classification. Polymorphic PTLD usually shows a heterogeneous population of small to medium-sized lymphocytes and plasma cells with a spectrum ranging from polymorphic lymphoid hyperplasia to Hodgkin-lymphoma-like to near monomorphic PTLD.[34]
Primary effusion PTLD is usually positive for B-cell markers (CD19, CD20, CD79a) and may express CD30 and CD43.[34,62] T-cell, Hodgkin lymphoma, high grade B cell lymphoma and null phenotype PTLDs have been reported occasionally. The T-cell PTLDs express pan-T cell markers (CD3, CD5, and CD43) and may express CD4, CD8, CD56, and CD30. The Hodgkin lymphoma PTLD may express CD15 and CD30 and virtually all are EBV positive.[34,63]
Anaplastic large cell lymphoma (ALCL)
ALCL is a T-cell lymphoma that accounts for 3% of adult non-Hodgkin lymphomas and 10–30% of childhood lymphoma. ALCL frequently involves lymph nodes and extranodal sites, including skin, bone marrow, soft tissue, lung, and liver. ALCL has been referred to as ‘Ki-1 lymphoma’ because of its Ki-1 (CD30) positive staining. Ki-1 was originally described as a marker for Hodgkin lymphoma, but it soon became apparent that it was not specific for Hodgkin lymphoma.[34,64]
ALCL has been reported in body fluids, including peritoneal fluid, pleural fluid, pericardial fluid, urine, and cerebrospinal fluid. These high-grade lymphomas may be confused with and cytomorphologically resemble a high-grade or undifferentiated carcinoma or sarcoma, including melanoma. One case of AIDS-related ALCL secondarily involving the pleural fluid was reported to resemble metastatic adenocarcinoma by forming gland-like structures.[3,64-66]
• ALCL characteristically consists of large, pleomorphic cells that often have a horseshoe-shaped or kidney-shaped nuclear configuration (also known as hallmark cells). Multinucleated forms may also be present, with nuclei arranged in a wreath-like pattern or sometimes resembling RS cells. The cells often have abundant cytoplasm, clumped chromatin, and one or more nucleoli. The inclusion-like nucleolus in the RS cell of Hodgkin lymphoma is uncommonly seen. Variants which have small lymphocyte and histiocytoid cytomorphology may be seen in 10% of ALCLs.
• ALCL is positive for CD30 (membrane and Golgi staining) in all cases. AKL-1 and EMA are positive in the majority of cases. Cytoplasmic and nuclear staining for ALK-1 is associated with the t(2;5) translocation of the NPM gene on chromosome 5 and the ALK gene on chromosome 2. Other translocations can be present, and in those cases, only cytoplasmic staining for ALK-1 will be present. The cells of ALCL are often CD3 negative, but other T-cell markers like CD4 and CD2 are often positive. CD15 (Leu-M1) and PAX5 are typically negative in ALCL and can help exclude Hodgkin lymphoma.[34,64] For differential diagnosis of other neoplasms that may also express CD30, please refer to Tables 1 and 2.
Adult T-cell leukemia/lymphoma (ATLL)
Adult T-cell leukemia/lymphoma (ATLL) is a mature T-cell neoplasm of post-thymic origin caused by human T-cell lymphotrophic virus, type 1 (HTLV-1), which was first isolated and identified in 1980. The disease is most frequent in Japan, the Caribbean basin, and West Africa, where the virus is endemic. It generally affects adults with generalized lymphadenopathy, hepatosplenomegaly, skin lesions, and hypercalcemia. This systemic disease commonly involves skin, lung, liver, spleen, gastrointestinal tract, and central nervous system.
Up to 15% of patients may have malignant pleural effusions, while 6% may have peritoneal effusions. A broad range of clinical presentations has led to the division of ATLL into four types: acute, lymphomatous, chronic, and smoldering. The acute and lymphomatous forms represent 55% and 20% of all ATLL cases, respectively. The acute form is the leukemic phase with high numbers of circulating neoplastic cells. The lymphomatous form is characterized by prominent lymphadenopathy without peripheral blood involvement. The chronic form is limited to the skin, while the smoldering form has <5% circulating neoplastic cells. The chronic and smoldering variants may progress to the acute type in 25% of cases.[34,67]
Cardiac involvement by ATLL occurs in 35–40% of cases. Autopsy reports describe neoplastic cells massively infiltrating the myocardium, involving the heart valves, and leading to aortic or mitral regurgitation; however, pericardial effusion presentation occurs only in rare cases. In general, cardiac involvement with ATLL occurs in patients with circulating leukemic cells, hepatosplenomegaly, generalized lymphadenopathy, or skin lesions. Recently, a case report described an ATLL patient presenting with a pericardial effusion and cardiac tamponade in the absence of skin lesions, lymphadenopathy, hepatosplenomegaly, peripheral blood or bone marrow involvement.[67]
• The cells of ATLL are medium-to large-sized with peculiar, polymorphic, markedly lobulated nuclei which assume a ‘flower-cell’ morphology, while the cells of chronic and smoldering variants are small to medium lymphocytes with mild to minimal atypia and indistinct nucleoli.[67] Variably prominent nucleoli are present.
The neoplastic lymphocytes are usually immunoreactive for CD2, CD3, CD4, CD5, and CD25 while lacking CD7 and CD8. Some cells may be positive for CD30 but are ALK negative. Integration of HTLV-1 virus DNA into ATLL tumor cell DNA is present in all cases. Serum antibodies to HTLV-1 may also be positive.[34,68]
Peripheral T-cell lymphoma, NOS
Peripheral T-cell lymphoma is an uncommon lymphoma accounting for 5–10% of NHL cases. It is more common in blacks than among whites.[40] Peripheral T-cell lymphoma commonly shows extranodal involvement (82% of cases) and in those cases, almost always involves the skin.[69] Peripheral T-cell lymphoma can involve any serous cavity. • It may present primarily as an effusion without evidence of tumor mass, but more commonly involves the serous cavities secondarily.
• Peripheral T-cell lymphomas can be challenging to diagnose in some cases because cytologic atypia may be minimal. In cases with obvious cytologic atypia, the neoplastic nature of the cells can be announced confidently without necessarily needing flow cytometry or gene rearrangement studies. In the other cases, loss of one or more pan-T-cell markers (CD5, CD3, CD43, CD7, and CD2) or a clonal T-cell receptor gene rearrangement pattern can be helpful in determining that the population of T-cells is neoplastic. The category of peripheral T-cell lymphoma NOS requires that the cells be negative for ALK-1.
Extranodal natural killer (NK) lymphoma
In rare cases, NK lymphoma may present as a peritoneal or pleural effusion.[70] The neoplasm characteristically involves the nasal cavity, and thus is named in the 2001 WHO classification as ‘extranodal NK/T-cell lymphoma, nasal type.’ Other names previously used for this tumor include angiocentric T-cell lymphoma and lethal midline granuloma. Besides the nasal cavity and nasopharynx, the sites of involvement include skin, soft tissue, testis, and gastrointestinal tract. NK lymphoma is more prevalent in Asia, Mexico, and Central and South America. Extranodal NK lymphoma outside the nasal cavity is highly aggressive and responds poorly to therapy.
The cytologic spectrum of the cells is highly variable. The cells may be small, medium, large, anaplastic, or a mixture. They may have small or inconspicuous nucleoli. The cytoplasm is pale and moderately abundant. With Giemsa staining, azurophilic granules can be identified within the cytoplasm. The tumor may also be accompanied by a mixture of inflammatory cells, including small lymphocytes, plasma cells, histiocytes, and eosinophils.
The typical immunophenotype of extranodal NK lymphoma is CD2, CD3c (cytoplasmic), and CD56 positive with lack of surface CD3. Other T-cell/NK antigens such as CD4, CD5, CD8, CD16, and CD57 are usually negative. Most cases are also positive for cytotoxic proteins such as granzyme B, TIA-1, and perforin. T-cell receptor gene rearrangement is characteristically negative. EBV can be detected in the tumor cells in most cases.[34]
Classical Hodgkin lymphoma
Hodgkin lymphoma accounts for approximately 30% of all lymphomas. Two major types of Hodgkin lymphoma are recognized: classical Hodgkin lymphoma and nodular lymphocyte predominant Hodgkin lymphoma. The latter type is more closely related to a large B-cell lymphoma, and will not be discussed here. Classical Hodgkin lymphoma (HL) is a distinct neoplasm from other B-cell lymphomas because of the famous RS cell. Four subtypes of classical Hodgkin lymphoma are recognized: nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted. The nodular sclerosis subtype accounts for 70% of all classical Hodgkin lymphoma cases. Nodular sclerosis is also the subtype most frequently reported to involve the mediastinum (80% of cases) and lung (10% of cases).[34]
Pleural effusion may be present in 20% of patients with Hodgkin lymphoma, due to obstruction of lymphatic drainage in the mediastinum or lung.[23] A diagnosis of a malignant effusion is only rarely made because (1) in the majority of patients the tumor remains subpleural, even in the face of a concurrent effusion, and (2) the neoplasm comprises relatively few RS cells, and their identification can be difficult. Application of immunohistochemical stains (see below) is required to increase the rate of identifying malignant effusion associated with Hodgkin lymphoma.
Cytomorphologically, classical Hodgkin lymphoma is composed of RS cells which have multilobated nuclei and a large, prominent, inclusion-like nucleolus. RS variants (sometimes called Hodgkin cells) are mononuclear, thus lacking the classic bilobed appearance of the classic RS cell. Except for the lymphocyte-depleted subtype, RS or RS-variant cells are not the predominant cell type constituting the neoplasm. In tissue, the predominant background cells in the majority of Hodgkin lymphoma cases are usually a mixture of reactive lymphocytes, plasma cells, eosinophils, and neutrophils which outnumber RS cells. • In serous fluid, lymphocytes usually predominate, but scattered to rare RS cells can be seen. By serous fluid cytology alone Hodgkin lymphoma can usually be suspected. RS cells must be identified and confirmed by immunophenotyping or tissue section histology before a definitive diagnosis can be made.
Immunophenotypically, RS cells characteristically express CD15, CD30 but not CD45. Although in the majority of cases, RS cells lack CD20 expression, heterogeneous CD20 expression may be observed up to 20% of CHL cases. PAX5 is also usually expressed by RS cells but the expression is weaker than the reactive B-cells in the background.
Even though the cells have a characteristic morphology and immunophenotype, numerous mimickers of Hodgkin lymphoma have been described. For instance, EBV-infected B cells outside a setting of classical Hodgkin lymphoma can have a RS-like appearance and can be CD30 and CD15 positive.[34] The immunostains for classical Hodgkin lymphoma are non-specific as CD30 marks activated lymphocytes, both T and B cells, while CD15 marks cells of myelomonocytic origin.[71] Diffuse large B-cell lymphoma, anaplastic large cell lymphoma, and primary effusion lymphoma can also be CD30 positive.[23,34,72]
Primary cardiac lymphoma
The WHO recognizes two definitions of primary cardiac lymphoma. The first defines it as an extranodal lymphoma that involves only the heart and/or pericardium.[52,73] However, the WHO also recognizes a less-restrictive definition in which the bulk of the tumor is arising in the heart and/or pericardium with small secondary lesions elsewhere. This less-restrictive definition has been explained by Ceresoli et al in the 1997 Cancer article which reviews primary cardiac lymphomas.[18] In this definition of primary cardiac lymphoma, the extracardiac site of involvement must be a single, asymptomatic focus in a patient who presents with cardiac lymphoma. Excluded from the primary cardiac lymphoma category are cardiac lymphomas with significant and symptomatic extracardiac sites of lymphoma. • Many cases of primary cardiac lymphoma reported in the literature in the past are now considered to be metastatic lymphoma (secondary cardiac lymphoma). Thus, the main criterion for this diagnosis is exclusion of cardiac involvement by a disseminated lymphoma elsewhere in the body.
Primary cardiac lymphoma accounts for 1.3% of cardiac malignancies and 0.5% of extranodal lymphoma. Most patients with it are immunocompetent males (male to female ratio of 3:1) with a median age of 62 years (range, 5–90 years). Clinical presentation is typically with chest pain, dyspnea, heart failure, arrhythmia, or syncope. In the majority of patients, the tumor arises in the right chambers of the heart. Primary cardiac lymphoma may present as an isolated mass (42% of cases), a mass and effusion (44% of cases), or an isolated effusion (12% of cases).[18] The mean length of survival after diagnosis is 7 months.[11,52]
When pericardial effusion is present in primary cardiac lymphoma, the lymphoma cells may be detected in the serous fluid in 67–88% of patients. In the absence of a pericardial effusion, however, a diagnostic cytologic sample is obtained in less than 20% of primary cardiac lymphomas. Diffuse large B-cell lymphoma is the most common form of lymphoma (80%) seen in primary cardiac lymphoma [Figure 5]. Burkitt lymphoma and low-grade ‘small’ cell lymphomas have also been reported. Only two cases of primary cardiac T-cell lymphoma have been reported in the literature to date.[74]
EOSINOPHILIC EFFUSIONS
Eosinophilic pleural effusions
• Eosinophilic pleural effusion is defined as such when >10% of the nucleated cells in the pleural fluid are eosinophils, have traditionally been associated with blood or air in the pleural space, as may occur with pulmonary infarction, spontaneous pneumothorax, prior chest surgery (e.g. coronary artery bypass graft [CABG] surgery), or even prior thoracentesis. [75] However, recent studies have shown that not all patients with prior CABG develop eosinophilic pleural effusions, RBC counts in pleural fluid do not correlate with eosinophilia, and patients with repeated thoracentesis may show decreasing eosinophil counts.[30,76,77] It is possible that pleural injury, the degree of pleural injury, and/or different individual responses to pleural injury may explain the eosinophilic pleural effusion.
Whatever the underlying mechanism, it has been shown by many studies that eosinophilic effusions are closely correlated with elevated interleukin-5 (IL-5) levels in the pleural fluid. IL-5 is a product of CD4+ T-lymphocytes.[76,78] Patients with eosinophilic pleural effusions often, but not always, have peripheral blood eosinophilia as well.[76] Eosinophilic pleuritis has been associated with many diseases, including tuberculosis, sarcoidosis, asbestosis, collagen vascular disease, malignancy, and drug allergy.[30,78] The most common cause of eosinophilic pleural effusion in patients from India is tuberculosis.[30] In the United States, the most common associations are ‘trauma’ (25%), congestive heart failure (14%), infection (8.5%), and idiopathic (8.5%). Tumor does not seem to be a common cause of eosinophilic pleural effusions.[79]
Eosinophilic peritoneal effusions
Eosinophilic ascites is relatively uncommon. Chronic peritoneal dialysis has been associated with peritoneal fluid eosinophilia.[9] Other various causes reported in the literature include eosinophilic gastroenteritis, Toxocara infection, adverse drug reactions, leukemia/lymphoma, and idiopathic hypereosinophilic syndrome.[80,81] In some reports, up to 75% of eosinophilic ascites cases occur in female patients with an average age of 40 years, a history of allergy (55%), peripheral blood eosinophilia (69%), history of gastrointestinal complaints, and eosinophilic infiltration of the bowel wall or serous membranes. These reports suggest eosinophilic gastroenteritis, both conventional type and subserous type, may be responsible for a large number of cases of eosinophilic ascites. The outcome in most of these cases of gastroenteritis is favorable with initiation of steroid therapy.[82-84] When no other cause can be found, the diagnosis of idiopathic hypereosinophilic syndrome may be appropriate. Lymphomas associated with an eosinophilic component include Hodgkin lymphoma, natural killer cell lymphoma, and T-cell lymphoma.
Eosinophilic pericardial effusions
Eosinophilic pericardial effusions are rare. The various causes reported in the literature include Churg–Strauss syndrome, Toxocara infection, Löffler syndrome, adverse drug reactions, and idiopathic hypereosinophilic syndrome.[85,86] Idiopathic hypereosinophilic syndrome is defined as the following: a peripheral blood absolute eosinophil count that exceeds 1500/μL for at least 6 months’ duration, exclusion of other known causes of eosinophilia, and organ system dysfunction. In a case of idiopathic hypereosinophilic syndrome with pericardial effusion, the fluid had a WBC count of 14 800/ ml, with 61% neutrophils, 23% eosinophils, and 15% monocytes.[85] • If an increased number of blasts is seen, or if evidence of clonality is shown (i.e. a recurring karyotypic abnormality), the diagnosis may be chronic myelogenous or chronic eosinophilic leukemia rather than idiopathic hypereosinophilic syndrome.[34] Hodgkin lymphoma is another common cause of eosinophilic pericardial effusions. If RS cells are seen, Hodgkin lymphoma should be considered.
NEUTROPHILIC EFFUSIONS
Reactive/benign neutrophilic effusions
Pulmonary embolism
The differential WBC count in patients with pulmonary embolism is more often predominated by neutrophils (60% of patients), but in 40% of patients, lymphocytes predominate. Greater than 10% eosinophils can be seen in up to 18% of effusions secondary to pulmonary embolism. The effusion characteristically is an exudate and frequently shows increased mesothelial cells and RBCs.[26]
Parapneumonic effusions
Parapneumonic effusions are associated with bacterial pneumonia, lung abscess, or bronchiectasis. Parapneumonic effusions are the most common cause of exudative pleural effusions in the USA and are the most common cause of neutrophilic pleural effusions. Empyema refers to a grossly purulent pleural effusion.[87]
Spontaneous bacterial peritonitis
Patients with cirrhosis and ascites are prone to develop spontaneous bacterial peritonitis. Multiple mechanisms have been hypothesized and it is likely that more than one factor may contribute to its development. One factor is the low content of protective proteins (opsonins) normally present in peritoneal fluid of a person with normal liver function. Another observation in rodent models with advanced cirrhosis is bacterial overgrowth in the intestine. The bacteria then leak into mesenteric lymphatics and enter mesenteric lymph nodes. From the lymph nodes, bacteria enter the systemic circulation. Uncleared bacteria in the circulating blood then leak from the Glisson’s capsule of the cirrhotic liver into the ascitic fluid.[88]
Spontaneous bacterial peritonitis is characterized by a turbid and purulent fluid. RBCs in the fluid are usually low in number, although a traumatic paracentesis may change this finding. Greater than 50% granulocytes or more than 250 granulocytes/μL should raise the index of suspicion for spontaneous bacterial peritonitis. The finding of >500/μL is virtually diagnostic of spontaneous bacterial peritonitis.[9,87] A Gram stain may show microorganisms and neutrophils in the fluid [Figure 6]. The most common organisms cultured in spontaneous bacterial peritonitis are Gram-negative enteric bacteria. Approximately 50% of cases are due to Escherichia coli or Klebsiella.[87,88]
- Neutrophils with intracellular bacteria (arrow) in neutrophilic peritonitis.
Infectious pericarditis
In most cases, the predominant cell type in infectious bacterial pericarditis is the neutrophil. The etiologic agent is most commonly Staphylococcus aureus. Gram stain and cultures will help make the diagnosis.
Systemic lupus erythematosus (SLE)
The characteristic cell in SLE is the so-called LE cell. The LE cells occur in a background of numerous neutrophils and RBCs, with scattered lymphocytes, macrophages, and mesothelial cells.[89] LE cells have been reported in the bone marrow, peripheral blood, cerebrospinal fluid, pleural, pericardial, and peritoneal fluids. LE cells are strongly associated with SLE but are not specific since they have been reported in patients taking certain medications (e.g. procainamide, hydralazine, and isoniazid) associated with an SLE-like syndrome. Additionally, they are present only in a minority of SLE cases.
LE cells represent phagocytes (usually neutrophils, but also monocytes) that have phagocytosed nuclei of other cells coated with circulating antinuclear antibodies. The phagocytosed and partially digested nuclear material is referred to as a ‘hematoxylin body’ because of blue staining with the PAP, Wright-Giemsa, and DQ stains Figure. The hematoxylin bodies may also occur free and in groups. In some cases, the hematoxylin body may push the phagocyte nucleus to the periphery, creating the appearance of a signet-ring cell seen in adenocarcinoma.
Neoplastic neutrophilic effusions
Extramedullary hematopoiesis/chronic idiopathic myelofibrosis
Extramedullary hematopoiesis is a common feature of chronic idiopathic myelofibrosis, which is a neoplastic stem cell disorder of the bone marrow resulting in fibrosis and extramedullary hematopoiesis in organs such as the spleen and liver. In some cases, portal hypertension resulting from liver infiltration by extramedullary hematopoietic cells can result in ascites. In other instances, the peritoneum itself may be involved by extramedullary hematopoiesis tissue, resulting in a reactive peritoneal serous effusion. Extramedullary hematopoiesis tissue in the pleura may similarly produce a pleural effusion.
Cytologic examination of the serous fluid will yield immature and maturing hematopoietic cells, including neutrophilic myelocytes, nucleated red cells, and megakaryocytes. • Megakaryocytes may be the most readily identifiable cells because of their large size and distinctive multilobated nucleus [Figure 7]. Megakaryocytes are positive for factor VIII-related antigen, CD41, and CD61.[34] In studies totaling almost 5000 pleural and peritoneal fluid samples, megakaryocytes were only found in five: three had chronic idiopathic myelofibrosis, one had chronic myelogenous leukemia, and one had lymphoma.[90] Thus, a diagnosis of chronic idiopathic myelofibrosis cannot be made solely on the finding of extramedullary hematopoiesis in the absence of a bone marrow biopsy, but it can be suggested in a comment as a frequent cause of extramedullary hematopoiesis.
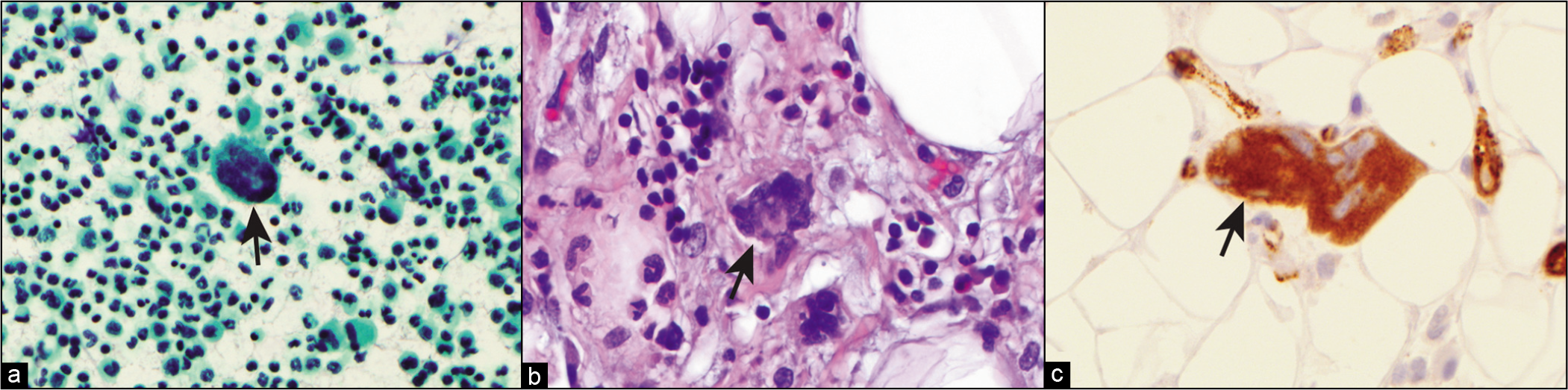
- (a) PAP-stained cytology smear from ascitic fluid showing megakaryocyte (arrow). (b) Tissue section from subsequent biopsy showing megakaryocytes in extramedullary hematopoiesis. (c) Tissue section with factor VIII immunohistochemistry staining of megakaryocyte.
Myeloid sarcoma
Primary presentation of myeloid sarcoma in effusions is extremely rare. Of seven cases reported in the literature, three occurred in the pleural cavity, three in the peritoneal cavity, and one in the pericardial cavity.[91] Myeloid sarcoma is a term chosen by the 2017 WHO classification to describe what has been previously termed granulocytic sarcoma or chloroma. Myeloid sarcoma is a rare tumor composed of myeloid (granulocytic, monocytic, megakaryocytic, erythroid) precursors that usually occurs with or heralds the development of acute myelogenous leukemia (AML) or rarely, chronic myelogenous leukemia, Myelodysplastic chronic myelogenous leukemia (CML), Myelodysplastic Syndrome (MDS), and myeloproliferative neoplasm (MPN). • There is no blast percentage cutoff that defines myeloid sarcoma, but instead the entity is recognized as a proliferation of immature and mature myeloid precursors, which differs from the trilineage proliferation of myeloid, megakaryocytic, and erythroid precursors seen in extramedullary hematopoiesis.
The cellular composition of myeloid sarcoma can vary depending on the type of leukemia and the degree of cell maturation and differentiation [Figure 8]. Thus, the cells in myeloid sarcoma and simultaneously occurring AML subtype may be identical. The prototypical immature myeloblast is medium in size (1.5–2 times the size of a resting lymphocyte) with scanty basophilic cytoplasm. The myeloblast nucleus has fine chromatin and several nucleoli. As the myeloblasts mature into promyelocytes, cytoplasmic azurophilic granules appear and accumulate. These abnormal granules may clump together forming Auer rods. Auer rods are most frequently seen in acute promyelocytic leukemia (M3) and acute myeloblastic leukemia with maturation (M2).
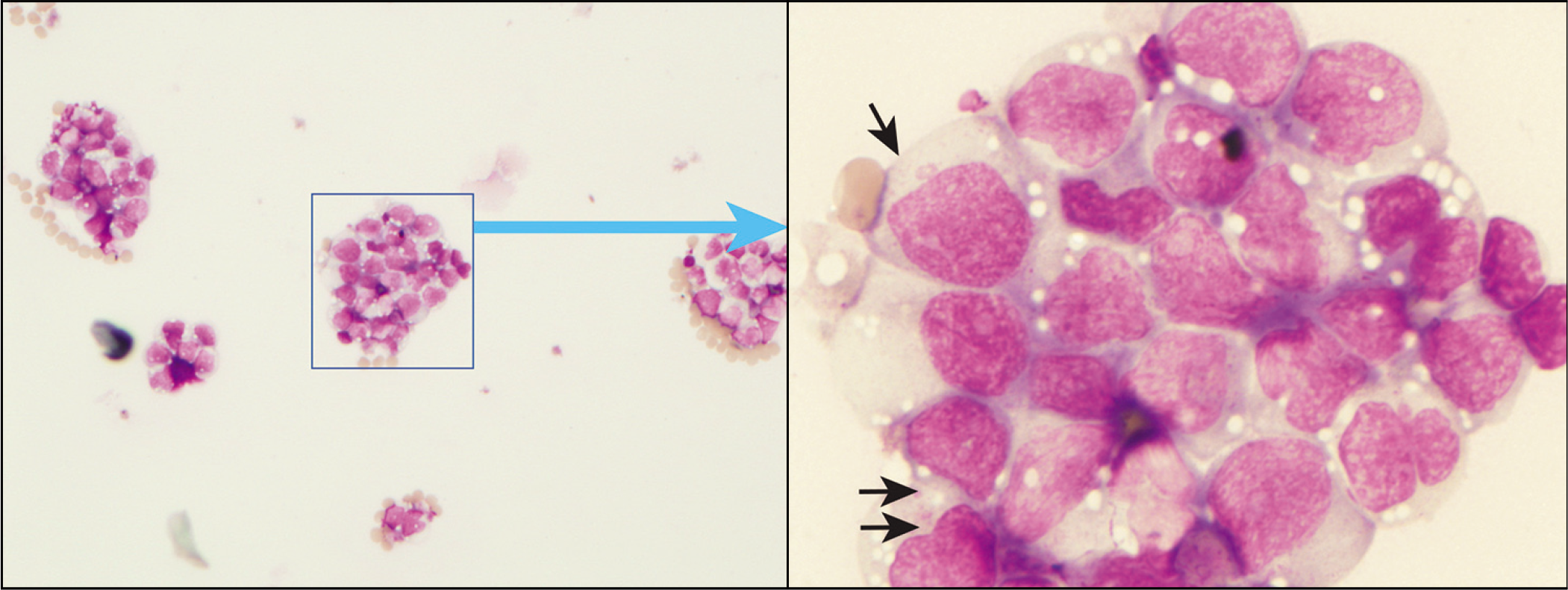
- Clusters of blasts and immature myeloid cells with cytoplasmic granules (arrows) and irregular nuclei in myeloid sarcoma.
• Flow cytometry would be very helpful in diagnosing these tumors since the possible combination of markers is more extensive than what is available by cytochemical or immunohistochemistry stains. Considering the most common types of AML are acute myelomonocytic leukemia (M4) and acute myeloblastic leukemia with maturation (M2), one can predict these cells should mark with CD45, CD34, CD117(c-kit), myeloperoxidase, and/or lysozyme. Acute myelomonocytic leukemia typically is positive for non-specific esterase (NSE). B-cell and T-cell markers are generally negative; however, the T-cell marker CD43 is commonly positive. Staining for lymphoid markers does not exclude a diagnosis of myeloid sarcoma. However, the 2017 WHO classification of acute myeloid leukemia is based on the presence of specific gene translocations and mutations.[34,91]
Chronic myelogenous leukemia (CML)
CML, a member of the category of chronic myeloproliferative diseases, is defined by the association with the Philadelphia chromosome t(9;22) and/or the BCR/ABL fusion gene. The initial major finding in CML is a neutrophilic leukocytosis. The neoplastic proliferation is primarily confined to the blood, bone marrow, spleen, and liver. However, extramedullary involvement can occur and does so most commonly during the accelerated (10–19% blasts) or blast phase (>20% myeloblasts or lymphoblasts) of CML. In very rare cases, CML has been reported to manifest as a pleural, pericardial, or peritoneal effusion. Case reports have described patients who showed resolution of the effusion after imatinib therapy.[92,93]
Cytomorphologically, CML is a neutrophilic proliferation. The abnormal neutrophils usually show decreased leukocyte alkaline phosphatase staining; however; this is not specific for CML. The cellular population may show an increased number and percentage of basophils. Eosinophils may be increased in some patients. Increased numbers of blasts are seen during the accelerated phase (10–19% blasts) and during the blast phase (>20% blasts). Cytogenetics and/or molecular studies are needed to make a definitive diagnosis.
HISTIOCYTIC EFFUSIONS
Rheumatoid arthritis
Rheumatoid arthritis patients may occasionally develop a pleural effusion (5% of patients). The pleura in these cases is involved with necrotizing granulomatous inflammation. The fluid characteristically is composed predominantly of histiocytes, some multinucleated, and scattered lymphocytes and granulocytes. Mesothelial cells are sparse. Abundant granular and irregular clumps of debris dominate the background.[32] The histiocytes in rheumatoid arthritis may have an epithelioid, spindled shape, mimicking squamous cells.[1]
Nodular histiocytic hyperplasia
Nodular histiocytic hyperplasia is a benign histiocytic lesion caused by mechanical irritation, inflammation, or tumor affecting the pleura. Effusions consist predominantly of bland histiocytes with background lymphocytes and mesothelial cells.[94] The histiocytic proliferation may simulate a mesothelial reaction or a malignant tumor. Histiocytes can often be distinguished from mesothelial cells by their lack of a peripheral cytoplasmic clearing-zone which corresponds to microvilli of mesothelial cells. In addition, CD68 and CD163 are positive in the histiocytes/ macrophages. The differential diagnosis may include diffuse large B-cell lymphoma, lymphoblastic lymphoma/leukemia, rhabdomyosarcoma, PNET/Ewing’s sarcoma, melanoma, and poorly-differentiated carcinoma.
IMMUNOPHENOTYPIC COMPARISON OF VARIOUS LYMPHOMAS/LEUKEMIAS [SEE TABLE 2]
CASE STUDIES
CASE 1
History
A 51-year-old male presented with nausea and abdominal pain. A gastrointestinal work-up, including gastric and intestinal biopsies, liver function tests, and hepatitis screening tests, was negative. Abdominal computed tomography (CT) did not show mass lesions, but a moderate amount of abdominal ascites was present. Paracentesis was performed, and fluid was sent for cell count and to cytopathology for cytologic examination.
Cytomorphology
Cytologic examination revealed a monomorphic dispersed population of medium to large cells with moderate amounts of basophilic cytoplasm with a few cytoplasmic vacuoles. The nuclei were round to slightly irregular with fine chromatin. Variably prominent nucleoli were identified. Cytoplasmic granules or Auer rods were not present.
Cytopathologic interpretation
Lymphoma, final characterization pending immunophenotyping [see Figure 4].
Immunophenotyping
Immunostains showed strong cytoplasmic staining in the tumor cells for CD30 and HHV-8. The tumor cells were nonimmunoreactive for CD45, CD20, CD10, CD3, CD4, CD5, CD34, MPO, lysozyme, melan-A, CD43, ALK-1, CD15, CD79a, CD138, cytokeratin, and EBER (EBV-ISH).
Flow cytometric analysis was limited due to low viability and cellularity but a small population of large cells weakly expressed CD20 and CD19. Immunoglobulin heavy chain and T-cell receptor gene rearrangement studies were negative for a clonal population. Real-time polymerase chain reaction (PCR) studies were positive for HHV-8 DNA and negative for EBV DNA.
Final interpretation
Primary effusion lymphoma.
Discussion
Primary effusion lymphoma can be a difficult diagnosis because often the typical B-cell neoplastic lymphoid markers are not positive, and in some cases CD3 may be positive. This may may lead to the interpretation as ‘undifferentiated malignant neoplasm’ unless HHV-8 and some B-cell lymphoid marker can be shown. A clinical history of HIV or immunosuppression may alert one to the possible diagnosis. The differential diagnosis in this case includes Burkitt lymphoma, diffuse large B-cell lymphoma, lymphoblastic lymphoma/leukemia, rhabdomyosarcoma, PNET/Ewing’s sarcoma, melanoma, and poorly-differentiated carcinoma.
CASE 2
History
A 6-year-old male presented to the physician with a history of constipation for several years. During the last 6 months, he had episodes of encopresis (involuntary defecation), which occurred twice a week. During the last week, his mother noticed increasing abdominal distention for which he was treated with stool softeners and enema. Over the next 2 days, his abdomen continued to increase in size. He only produced small amounts of liquid stool after treatment for constipation. He also had an episode of non-bloody emesis. Mother also reported that the child had had breathing difficulty for the last week. A subsequent CT scan showed an abdominal mass, thickened bowel wall, periportal and mesenteric lymphadenopathy, ascites, bilateral pleural effusions, hydronephrosis, and bilateral renal parenchymal enhancements. Lymphoma was strongly suspected and fluid from the pleural and abdomen was sent for cytologic examination and flow cytometry.
Cytomorphology
Cytologic examination revealed a monomorphic population of medium-sized lymphoid cells with a small amount of cytoplasm with numerous cytoplasmic vacuoles identified in many of the cells. Mitoses were readily identified. Macrophages with phagocytosed apoptotic material were also present.
Cytopathologic interpretation
Lymphoma, favor Burkitt lymphoma. Final pending immunophenotyping and fluorescence in-situ hybridization (FISH) for MYC rearrangement [Figure 2].
Flow cytometry
Flow cytometry showed a population of medium-sized lymphocytes with lambda light-chain restriction. The cells co-expressed CD45, CD10, CD20, and CD43. They were negative for CD5, CD38, CD56, and TdT.
FISH showed MYC rearrangement associated with t(8;14).
Final interpretation
Burkitt lymphoma.
Discussion
The cytologic and immunophenotypic features of Burkitt lymphoma are classic in this case. The patient’s age and clinical history are also typical. This tumor is rapidly growing and may present with a history suggestive of sudden rapid growth. The MYC translocation in this case is diagnostic and rules out other possible lymphomas that may be CD10 positive. The differential diagnosis of other CD10 positive B-cell lymphomas includes diffuse large B-cell lymphoma, follicular lymphoma, and lymphoblastic lymphoma.
Acknowledgment
Author of this review and editors of CMAS #2 series thank the initial authors (Steven R Sanchez, MD and Chung-Che (Jeff) Chang, MD, PhD) for all their efforts for the first edition material on which the current review (as chapter #12 in the final CMAS #2) is based.
Author thanks Andrew Kumar, MD (Resident, Wayne State University School of Medicine, Detroit, MI, USA) and Janavi Kolpekwar for copy-editing support.
ABBREVIATIONS (IN ALPHABETIC ORDER)
AIDS - Acquired immune deficiency syndrome
ALL - Acute lymphoblastic leukemia
ALCL - anaplastic large cell lymphoma
ALL/LBL - precursor T- or B- lymphoblastic leukemia/ lymphoma
AMLs - Acute myelogenous leukemias
ATLL - Adult T-cell leukemia/lymphoma
BL - Burkitt lymphoma
CABG - Coronary artery bypass graft
CLL - Chronic lymphocytic leukemia
CML - Chronic myelogenous leukemia
CT - Computed tomography
DLBCL - Diffuse large B-cell lymphoma
DQ - Diff-Quik
EBV - Epstein -Barr virus
FISH - Fluorescence in-situ hybridization
FL - Follicular lymphoma
FNAs - Fine needle aspirations
HHV-8 - Human herpes virus 8
HIV - Human immunodeficiency virus
HL - classical Hodgkin lymphoma
HTLV-1 - Human T-cell lymphotrophic virus, type 1
IL-5 - Interleukin-5
LE - Lupus erythematosus
LPL - lymphoplasmacytic lymphoma
MALT lymphoma - Mucosa-associated lymphoid tissue lymphoma
MCL - Mantle cell lymphoma
MZL - Marginal zone lymphoma
NHLs - non-Hodgkin lymphomas
NKL - Natural killer lymphoma
NLPHL - Nodular lymphocyte predominant Hodgkin lymphoma
NSE - Non-specific esterase
PAP – Papanicolaou
PCM - Plasma cell myeloma (multiple myeloma)
PTLD - Post-transplant lymphoproliferative disorder
RBCs - Red blood cells
RS - Reed -Sternberg cells
SLE - Systemic lupus erythematosus
SLL - Small lymphocytic lymphoma
TdT - Terminal deoxynucleotidyl transferase
WBC- White blood cell
References
- Lymphoid cell aggregates: a useful clue in the fine-needle aspiration diagnosis of follicular lymphomas. Diagn Cytopathol. 1997;17:467-71.
- [CrossRef] [Google Scholar]
- Secondary pleural involvement by an AIDS-related anaplastic large cell (CD30+) lymphoma simulating metastatic adenocarcinoma. Diagn Cytopathol. 1998;18:113-7.
- [CrossRef] [Google Scholar]
- Effusion cytology of malignant melanoma. A morphologic and immunocytochemical analysis including application of the MART-1 antibody. Cancer. 1997;81:57-63.
- [CrossRef] [Google Scholar]
- Cytology of fluids from pleural, peritoneal and pericardial cavities in children. A comprehensive survey. Acta Cytol. 1994;38:209-17.
- [Google Scholar]
- Collection and processing of effusion fluids for cytopathologic evaluation. CytoJournal. 2022;19:5.
- [CrossRef] [Google Scholar]
- Cell-blocks and hematolymphoid lesions. CytoJournal. 2021;18:7.
- [CrossRef] [PubMed] [Google Scholar]
- Cell-blocks and other ancillary studies (including molecular pathology and proteomics) CytoJournal. 2021;18:4.
- [CrossRef] [PubMed] [Google Scholar]
- The cytologic diagnosis of malignant neoplasms in pleural and peritoneal effusions. Acta Cytol. 1987;31:85-97.
- [Google Scholar]
- Universities Associated for Research and Education in Pathology In: Non-neoplastic disorders of the lower respiratory tract. Washington, DC: American Registry of Pathology; Armed Forces Institute of Pathology; 2002.
- [Google Scholar]
- The etiology of pleural effusions in an area with high incidence of tuberculosis. Chest. 1996;109:158-62.
- [CrossRef] [PubMed] [Google Scholar]
- Cytology of pleural, peritoneal and pericardial fluids in children. A 40-year summary. Acta Cytol. 1997;41:467-73.
- [CrossRef] [PubMed] [Google Scholar]
- Normal volume and cellular contents of pleural fluid. Paediatr Respir Rev. 2004;5(Suppl A):S201-3.
- [CrossRef] [Google Scholar]
- Volume and cellular content of normal pleural fluid in humans examined by pleural lavage. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1023-6.
- [CrossRef] [PubMed] [Google Scholar]
- The composition of normal pericardial fluid and its implications for diagnosing pericardial effusions. Am J Med. 2005;118:636-40.
- [CrossRef] [PubMed] [Google Scholar]
- The usefulness of a panel of immunostains in the diagnosis and differentiation of metastatic malignancies in pericardial effusions. Cytopathology. 2000;11:312-21.
- [CrossRef] [PubMed] [Google Scholar]
- Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer. 1997;80:1497-506.
- [CrossRef] [Google Scholar]
- Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore). 2003;82:385-91.
- [CrossRef] [PubMed] [Google Scholar]
- Application of molecular genetics to the diagnosis of lymphoid-rich effusions: study of 95 cases with concomitant immunophenotyping. Diagn Cytopathol. 2002;27:90-5.
- [CrossRef] [PubMed] [Google Scholar]
- Transudative malignant pleural effusions: prevalence and mechanisms. South Med J. 1998;91:23-6.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural effusion in patients with non-Hodgkin lymphoma: a case-controlled study. Cancer. 1998;83:1607-11.
- [CrossRef] [Google Scholar]
- Pleural fluid cytology of Hodgkin disease: cytomorphologic features and the value of immunohistochemical studies. Diagn Cytopathol. 2000;22:21-4.
- [CrossRef] [Google Scholar]
- The lipoprotein profile of chylous and nonchylous pleural effusions. Mayo Clin Proc. 1980;55:700-4.
- [Google Scholar]
- Exudative effusions in congestive heart failure. Chest. 2002;122:1518-23.
- [CrossRef] [PubMed] [Google Scholar]
- Biochemical and cytologic characteristics of pleural effusions secondary to pulmonary embolism. Chest. 2002;121:465-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cytologic diagnosis of syphilitic pleuritis: a case report. Diagn Cytopathol. 1997;16:35-8.
- [CrossRef] [Google Scholar]
- Cytologically malignant lymphoid pericardial effusion with benign clinical outcome. Swiss Med Wkly. 2005;135:377-81.
- [Google Scholar]
- Pleural effusions in hospitalized patients receiving long-term hemodialysis. Chest. 1995;108:470-4.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic value of eosinophils in pleural effusion: a prospective study of 26 cases. Diagn Cytopathol. 2003;28:96-9.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural effusions in the medical ICU: prevalence, causes, and clinical implications. Chest. 1997;111:1018-23.
- [CrossRef] [PubMed] [Google Scholar]
- Cytology: Diagnostic Principles and Clinical Correlates (2nd edn). New York: WB Saunders; 2003.
- [Google Scholar]
- Atlas of Diagnostic Cytopathology. (2nd edn). Philadelphia: WB Saunders; 2004. p. :856. xx
- [Google Scholar]
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (4th ed). Lyon, France: International Agency for Research on Cancer; 2017.
- [Google Scholar]
- International Agency for Research on Cancer. In: World Cancer Report. Lyon: IARC Press; 2003.
- [CrossRef] [Google Scholar]
- Pulmonary complications in chronic lymphocytic leukemia. Cancer. 2003;98:1912-7.
- [CrossRef] [PubMed] [Google Scholar]
- Cytology and immunophenotyping of lowand intermediate-grade B-cell non-Hodgkin lymphomas with a predominant small-cell component: a study of 56 cases. Diagn Cytopathol. 2001;24:90-7.
- [CrossRef] [Google Scholar]
- Immunoglobulin gene rearrangement in T-cell-rich reactive pleural effusion of a patient with B-cell chronic lymphocytic leukemia. Acta Haematol. 1996;96:41-4.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural involvement in B-cell chronic lymphocytic leukemia associated with a T-cell-rich 'reactive' pleural effusion. Am Rev Respir Dis. 1986;134:172-4.
- [Google Scholar]
- Cancer surveillance series: non-Hodgkin lymphoma incidence by histologic subtype in the United States from 1978 through 1995. J Natl Cancer Inst. 2000;92:1240-51.
- [CrossRef] [PubMed] [Google Scholar]
- Bethesda Handbook of Clinical Hematology Philadelphia: Lippincott, Williams & Wilkins; 2005.
- [Google Scholar]
- Cytologic findings of marginal zone lymphoma. Cancer. 2003;99:301-9.
- [CrossRef] [PubMed] [Google Scholar]
- Case report: mantle cell lymphoma, prolymphocytoid variant, with leukostasis syndrome. Mod Pathol. 2004;17:879-83.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebrospinal fluid involvement in Mantle cell lymphoma. Mod Pathol. 2002;15:1073-9.
- [CrossRef] [PubMed] [Google Scholar]
- Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia: one disease or three? Am J Clin Pathol. 2001;116:799-801.
- [CrossRef] [Google Scholar]
- Waldenstrom macroglobulinemia involving extramedullary sites: morphologic and immunophenotypic findings in 44 patients. Am J Surg Pathol. 2003;27:1104-13.
- [CrossRef] [PubMed] [Google Scholar]
- Neoplastic Hematopathology, 2nd edn Philadelphia: Lippincott, Williams & Wilkins; 2001.
- [Google Scholar]
- Effusion cytology in Burkitt's lymphoma. Diagn Cytopathol. 1995;12:3-7.
- [CrossRef] [PubMed] [Google Scholar]
- Hematologic malignancies with primary retroperitoneal presentation: clinicopathologic study of 32 cases. Arch Pathol Lab Med. 2005;129:655-60.
- [CrossRef] [PubMed] [Google Scholar]
- Ovarian non-Hodgkin lymphoma: a clinicopathologic study of eight primary cases. Mod Pathol. 2001;14:1093-9.
- [CrossRef] [PubMed] [Google Scholar]
- Human herpesvirus 8 (HHV-8)-associated peritoneal primary effusion lymphoma (PEL) in two HIV-negative elderly patients. Am J Hematol. 2004;76:88-91.
- [CrossRef] [PubMed] [Google Scholar]
- International Agency for Research on Cancer, International Association for the Study of Lung Cancer, International Academy of Pathology, Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart Lyon: IARC Press; 2004.
- [Google Scholar]
- HHV-8 associated T-cell lymphoma in a lymph node with concurrent peritoneal effusion in an HIV-positive man. Am J Surg Pathol. 2005;9:647-52.
- [CrossRef] [PubMed] [Google Scholar]
- Human herpesvirus 8associated solid lymphomas that occur in AIDS patients take anaplastic large cell morphology. Mod Pathol. 2000;13:77-85.
- [CrossRef] [PubMed] [Google Scholar]
- Primary effusion lymphoma: cytopathologic diagnosis using in situ molecular genetic analysis for human herpesvirus 8. Mod Pathol. 2002;15:944-50.
- [CrossRef] [PubMed] [Google Scholar]
- Primary effusion lymphoma of the pericardial cavity carrying t(1;22)(q21;q11) and t(14;17)(q32;q23) Cancer Genet Cytogenet. 2005;156:49-53.
- [CrossRef] [PubMed] [Google Scholar]
- HIV and HHV8 negative primary effusion lymphoma in a patient with hepatitis C virus-related liver cirrhosis. Leuk Lymphoma. 2003;44:1811-4.
- [CrossRef] [PubMed] [Google Scholar]
- Malignant pleural effusion of multiple myeloma: prognostic factors and outcome. Leuk Lymphoma. 2005;46:1137-42.
- [CrossRef] [PubMed] [Google Scholar]
- Multiple myeloma involving the porta hepatis and peritoneum causing biliary obstruction and malignant ascites. Dig Dis Sci. 2005;50:1068-71.
- [CrossRef] [PubMed] [Google Scholar]
- Cytology and flow cytometry of malignant effusions of multiple myeloma. Diagn Cytopathol. 2000;22:147-51.
- [CrossRef] [Google Scholar]
- Primary pleural effusion posttransplant lymphoproliferative disorder: distinction from secondary involvement and effusion lymphoma. Diagn Cytopathol. 2001;25:50-3.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural effusions in hematologic malignancies. Chest. 2004;125:1546-55.
- [CrossRef] [PubMed] [Google Scholar]
- Cytologic diagnosis of Ki-1 lymphoma in pleural and peritoneal effusions: a case report. Diagn Cytopathol. 1997;17:134-7.
- [CrossRef] [Google Scholar]
- Cytologic evidence for disseminated immunoblastic lymphoma. Am J Clin Pathol. 1981;75:621-5.
- [CrossRef] [PubMed] [Google Scholar]
- Value of lymphocyte marker studies in diagnostic cytopathology. Acta Cytol. 1987;31:453-9.
- [Google Scholar]
- Pericardial effusion: a rare presentation of adult T-cell leukemia/lymphoma. Am J Hematol. 2004;77:381-3.
- [CrossRef] [PubMed] [Google Scholar]
- Adult T-cell leukemia/lymphoma: a cytopathologic, immunocytochemical, and flow cytometric study. Cancer. 2002;96:110-6.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of non-Hodgkin lymphoma (NHL): trends, geographic distribution, and etiology. Ann Hematol. 2005;84:1-2.
- [CrossRef] [PubMed] [Google Scholar]
- Body cavity-based presentation of natural killer cell lymphoma. Leuk Lymphoma. 2005;46:293-6.
- [CrossRef] [PubMed] [Google Scholar]
- Immunocytochemistry in effusion cytology: a contemporary review. Cancer. 2001;93:293-308.
- [CrossRef] [PubMed] [Google Scholar]
- CD-30(Ki-1) positive anaplastic large cell lymphoma in a pleural effusion. A case report with diagnosis by cytomorphologic and immunocytochemical studies. Acta Cytol. 1999;43:498-502.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenesis of the eosinophilic pleural effusions. Curr Opin Pulm Med. 2004;10:289-93.
- [CrossRef] [PubMed] [Google Scholar]
- Eotaxin-3 and interleukin-5 pleural fluid levels are associated with pleural fluid eosinophilia in post-coronary artery bypass grafting pleural effusions. Chest. 2005;127:2094-100.
- [CrossRef] [PubMed] [Google Scholar]
- Etiology and prognostic significance of eosinophilic pleural effusions. A prospective study. Chest. 1996;110:1271-4.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural fluid levels of interleukin-5 and eosinophils are closely correlated. Chest. 2002;122:576-80.
- [CrossRef] [PubMed] [Google Scholar]
- Eosinophilic pleural effusion: a review of 36 cases. Respiration. 1985;48:73-6.
- [CrossRef] [PubMed] [Google Scholar]
- Eosinophilic gastroenteritis in a young girl-long term remission under Montelukast. BMC Gastroenterol. 2005;5:24.
- [CrossRef] [PubMed] [Google Scholar]
- Toxocara canis infection presenting as eosinophilic ascites and gastroenteritis. Dig Dis Sci. 1994;39:1370-72.
- [CrossRef] [PubMed] [Google Scholar]
- [Eosinophilic ascites, 2 new case reports] Rev Med Interne. 1992;13:446-8.
- [CrossRef] [Google Scholar]
- Eosinophilic gastroenteritis and ascites. J Clin Gastroenterol. 1981;3:371-3.
- [CrossRef] [PubMed] [Google Scholar]
- [Eosinophilia and ascites as an expression of a subserous form of eosinophilic gastroenteritis] Rev Clin Esp. 1992;91:30-4.
- [Google Scholar]
- Eosinophilic pericarditis: a rare complication of idiopathic hypereosinophilic syndrome in a child. Pediatr Cardiol. 2004;5:690-2.
- [CrossRef] [PubMed] [Google Scholar]
- Eosinophilic pericardial effusion in Churg-Strauss syndrome. Respir Med. 2005;9:377-9.
- [CrossRef] [PubMed] [Google Scholar]
- Harrison's Principles of Internal Medicine, 14th edn New York: McGraw-Hill, Health Professions Division; 1998.
- [Google Scholar]
- Diagnosis of systemic lupus erythematosus in an elderly male by pericardial fluid cytology: a case report. Diagn Cytopathol. 1998;18:346-8.
- [CrossRef] [Google Scholar]
- Ascites and pleural effusion secondary to extramedullary hematopoiesis. Am J Med Sci. 1999;318:286-8.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of granulocytic sarcoma on pleural effusion cytology: report of a case. Diagn Cytopathol. 2004;31:126-8.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic myeloid leukemia presenting with extramedullary disease as massive ascites responding to imatinib mesylate. Leuk Lymphoma. 2005;46:1097-9.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural-pericardic effusion as uncommon complication in CML patients treated with Imatinib. Eur J Haematol. 2005;74:89-90.
- [CrossRef] [Google Scholar]
- Cytologic clue of so-called nodular histiocytic hyperplasia of the pleura. Diagn Cytopathol. 2001;24:256-9.
- [CrossRef] [PubMed] [Google Scholar]




