


Translate this page into:
Silencing fatty acid-binding protein 4 improved sepsis-induced myocardial dysfunction through anti-apoptotic and antioxidant effects by mammalian target of rapamycin signaling pathway

*Corresponding author: Wei Chen, Department of Emergency, Hangzhou Xixi Hospital, Hangzhou, China. cw198512@163.com
-
Received: ,
Accepted: ,
How to cite this article: Qiu Z, Zhou K, Qi Q, Chen W. Silencing fatty acid-binding protein 4 improved sepsis-induced myocardial dysfunction through anti-apoptotic and antioxidant effects by mammalian target of rapamycin signaling pathway. CytoJournal. 2025;22:8. doi: 10.25259/Cytojournal_157_2024
Abstract
Objective:
One of the main complications of sepsis that is linked to poor clinical outcomes and high mortality is sepsis-induced myocardial dysfunction (SIMD). Fatty acid-binding protein 4 (FABP4) is a protein that is expressed in macrophages and adipose tissue and is involved in inflammation and apoptosis in various pathological processes. The purpose of this study was to investigate the role of FABP4 in SIMD.
Material and Methods:
The H9c2 cell model of myocardial dysfunction induced by septicemia was established by lipopolysaccharide (LPS). Measurements of cell viability, apoptosis, reactive oxygen species levels, mitochondrial activity, and proinflammatory factor expression were used to assess FABP4’s involvement in SIMD. In addition, the expression level of key proteins in the mammalian target of rapamycin (mTOR) signaling pathway was analyzed using Western blot. Finally, the combination of AZD-8055 further demonstrated the possibility of mTOR as a therapeutic target for SIMD.
Results:
Silencing FABP4 expression drastically increased H9c2 cell viability and mitochondrial function. In addition, by upregulating B-cell lymphoma-2 (Bcl-2) and downregulating Bcl-2 associated X protein, FABP4 silencing improved LPS-induced anti-apoptosis of H9c2 cells. Finally, silencing FABP4 alleviated SIMD through the mTOR signaling pathway. However, the therapeutic effect was inhibited when FABP4 silencing was combined with the mTOR inhibitor AZD-8055.
Conclusion:
Silencing FABP4 alleviates LPS-induced inflammatory response and apoptosis in H9c2 cells and enhances mitochondrial function through the mTOR signaling pathway.
Keywords
Fatty acid-binding protein 4
Mammalian target of rapamycin
Sepsis

INTRODUCTION
Sepsis-induced myocardial dysfunction (SIMD) refers to widespread acute myocardial damage caused by septicemia.[1] Over the past few decades, SIMD has become a major focus of research,[2] and its associated mortality has substantially increased.[3] Studies have shown that mitochondrial dysfunction and the production of reactive oxygen species (ROS) are linked to SIMD.[4] In addition, during the development of SIMD, immunity function becomes dysregulated,[5] which can further lead to tissue damage and molecular imbalances, ultimately resulting in organ dysfunction and failure.[6] Inflammatory cytokines are secreted in large quantities due to this immune dysfunction[7,8] and lymphocyte apoptosis is accelerated.[9,10]
The carrier protein for fatty acids in cells is called fatty acid-binding protein (FABP), which is essential for the cell’s utilization of fatty acids.[11] FABP4 is one of the FABPs and is most abundant in the heart and skeletal muscle.[12] FABP4 is well known for its detrimental effects in the development of insulin resistance,[13] diabetes,[14] gestational diabetes,[15] and metabolic syndrome.[16] Studies have shown that FABP4 knockout animal models exhibit an alleviating effect on insulin resistance. Following this discovery, FABP4 inhibitors became a major focus of research.[17] FABP4 is particularly associated with atherosclerosis and cardiovascular disease, with FABP4 deficiency playing a protective role in the early and late stages of atherosclerosis. Considering these findings, treating sepsis-induced cardiac dysfunction may benefit from targeting FABP4.
In addition, a study on in vivo pregnancy health has shown that inhibiting mammalian target of rapamycin (mTOR) signaling reduces FABP4 expression in the membrane and inhibits embryonic absorption.[18] However, a substantial amount of data also indicates that SIMD improves through the mTOR signaling pathway, revealing that mTOR may be a potential downstream target for SIMD treatment.[19] Therefore, this study first measured the level of FABP4 in H9c2 cells and then silenced FABP4 to further explore its possible role in SIMD. This study investigated the relationship between the mTOR signaling pathway and FABP4 by examining H9c2 cell viability, mitochondrial function, and anti-apoptotic capability.
MATERIAL AND METHODS
Cell culture
H9c2 rat embryonic cardiomyoblasts (BFN60804388, ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum (S9020, Solarbio, Beijing, China) and antibiotics (P7630, Solarbio, Beijing, China). The culture conditions were 37°C and 5% carbon dioxide. When the fusion rate of cells reached 70–80% in a 6-well culture dish, the cells were treated with 10 μg/mL LPS for 12 h,[20] establishing the cardiac injury cell model. Short tandem repeat analysis showed that the cells were produced from their parental cells and that they were clear of mycoplasma.
Cells were transfected using Lipofectamine 2000. FABP4 small interfering RNA (siRNA, si-FABP4): 5'-CCGAGAUUUCCUUCAAACU-3'. The experimental groups included the following: Control, lipopolysaccharide (LPS), si-NC (LPS + negative control to FABP4 siRNA), siFABPA (FABP4 siRNA + LPS), and si-FABP4 + AZD-8055 (LPS + FABP4 siRNA + 20 nM AZD-8055). The mTOR inhibitor AZD-8055 (No. 16978) was procured from Cayman Chemical (Ann Arbor, MI, USA).
Cell viability assay
The layered double hydroxides (LDHs) cytotoxicity assay kit (BC0685, Solarbio, Beijing, China) and the cell counting kit-8 (CCK-8, CA1210, Solarbio, Beijing, China) were used to evaluate the vitality of H9c2 cells. Cells were grown at a density of 5 × 103 cells/well in 96-well plates, and they were incubated for the entire night. After adding 10 μL of CCK-8 solution to each well and incubating the cells for 1.5 h at 37°C, the viability of the cells was evaluated. Absorbance was measured at 450 nm using an enzyme-labeled instrument (Multiskan SkyHigh, Thermo Fisher Scientific, Waltham, MA, USA).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining
Apoptosis in H9c2 cells was determined using the TUNEL kit (T2130, Solarbio, Beijing, China). After being cultured in 12-well plates, the cells were washed twice with phosphate buffer saline (P1010, Solarbio, Beijing, China) and then fixed in 4% paraformaldehyde for 25 min. 50 μL of TdT solution and 450 μL of fluorescein-labeled 2’-deoxyuridine 5’-triphosphate solution were added for staining after a 10-min soak in 1% Triton X-100. The staining process was carried out in the dark for 60 min. After a 15-min 4’,6-diamidino-2-phenylindole staining procedure, positive cells were observed and analyzed under fluorescence microscope (CX41-32RFL, Olympus Corporation, Tokyo, Japan).
Mitochondrial membrane Potential (MMP) (ΔΨm)
H9c2 cells were labeled with the fluorescent dye 5,5',6,6'-Tetrachloro-1,1',3,3'-tetraethyl-imidacarbocyanine (JC-1, M8650, Solarbio, Beijing, China) to evaluate the MMP. JC-1 was incubated for 30 min in the dark, then observed and analyzed using a fluorescence microscope. The ratio of red to normal mitochondrial potential and green to impaired mitochondrial potential was used to evaluate MMP.
ROS assay
ROS levels in H9c2 cells were measured using a ROS assay kit (CA1410, Solarbio, Beijing, China). H9c2 cells were incubated with LPS in a 6-well plate for 12 h, followed by the addition of 10 μL of 2`,7`-dichlorodihydrofluorescein diacetate at 37°C, protected from light. The relative fluorescence intensity was analyzed using a fluorescence microscope (CX41-32RFL, Olympus Corporation, Tokyo, Japan) and quantified with Image J (v1.8.0.345, National Institutes of Health, Bethesda, MD, USA).
Adenosine triphosphate (ATP) assay
The ATP levels in H9c2 cells were measured using an ATP kit (SP13572, Saipei Biotechnology Co., Ltd, Hubei, China). First, the cells were lysed, centrifuged at 4°C 12,000 g for 5 min, and the supernatant was collected. The test solution was added to each well of a 96-well plate and incubated at room temperature for 10 min, after which the supernatant was added. The optical density (OD) values of samples in 96-well plates were analyzed at 450 nm by enzyme-labeling apparatus (Multiskan SkyHigh, Thermo Fisher Scientific, Waltham, MA, USA).
Enzyme-linked immunosorbent assay (ELISA)
The expression levels of interleukin (IL)-1β (SEKH-002, Solarbio, Beijing, China) and tumor necrosis factor α (TNF-α) (SEKH-0047, Solarbio, Beijing, China) in H9c2 cells were determined using ELISA kits. After centrifuging the cells at 12,000 g and 4°C, the supernatant was gathered. Each well received 50 μL of the test sample and 50 μL of biotin, which were then incubated for 1 h at 37°C. After shaking the liquid in the hole for 30 s, additional washing liquid was incorporated, and the plate was shaken again. This step was repeated 3 times. The plate was then incubated at 37°C for 15 min without light after 50 μL of substrates A and B were added to each well. A final addition was 50 μL of termination solution. The OD value of each well was measured at 450 nm using an enzyme-labeled instrument.
Quantitative real-time–polymerase chain reaction (qRT–PCR)
Total ribonucleic acid (RNA) was extracted using TRIzol reagent (15596026CN, Thermo Fisher Scientific, Waltham, MA, USA) and quantified with the plate reader-based μDropTM Plate. Reverse transcription was performed using the RevertAid Reverse Transcription Kit (K1691, Thermo Fisher Scientific, Waltham, MA, USA). Primers [Table 1] and iQTM SYBR Green Supermix (1708880, Bio-RAD, Hercules, CA, USA) were used to prepare the quantitative polymerase chain reaction (qPCR) solution, which was then run on a Bio Rad qPCR apparatus (CFX Opus 96, Bio-RAD, Hercules, CA, USA). The obtained data were compared and interpreted relative to the reference gene (Glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) to assess gene expression differences between samples, and gene expression was quantified using the 2−ΔΔCT method.
| Primers | Primes sequences (5’-3’) |
|---|---|
| FABP4 forward | TGGGCCAGGAATTTGACGA |
| FABP4 reversed | CATTTCTGCACATGTACCAGGACAC |
| TNF-α forward | CATCTTCTCAAAATTCGAGTGACAA |
| TNF-α reversed | TGGGAGTAGACACAAGGTACAACCC |
| IL-1β forward | TGAACTGAAAGCTCTCCACCT |
| IL-1β reversed | ACTGGGCAGACTCAAATTCCA |
| GAPDH forward | GCACCGTCAAGGCTGAGAAC |
| GAPDH reversed | TGGTGAAGACGCCAGTGGA |
FABP4: Fatty acid-binding protein 4, TNF-α: Tumor necrosis factor-α, IL: Interleukin, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, A: Adenine, C: Cytosine, G: Guanine, T: Thymine
Immunofluorescent staining
H9c2 cells were immobilized on slides with 4% paraformaldehyde, then blocked with 2% goat serum (C0265, Beyotime Biotechnology, Shanghai, China) for 1 h after impregnation with 0.5% Triton X-100 for 20 min. Confocal microscopy was used to analyze primary antibodies that were incubated and specific secondary antibodies (1:1000, ab150077, AB105113, and ab150075) that were fluorescein-labeled.
The primary antibodies include the following: Inducible nitric oxide synthase (iNOS) (1:100, ab1789458), Superoxide dismutase 2 (SOD2) (1:100, ab13534), B-cell lymphoma-2 (Bcl-2) (1:100, ab194583), and Bcl-2 associated X protein (BAX) (1:100, ab216494). These antibodies were obtained from Abcam, Cambridge, MA, USA.
Western blot
Total protein content was determined using a bicinchoninic acid assay kit (PC0020, Solarbio, Beijing, China) after cells were lysed with radioimmunoprecipitation assay (R0010, Solarbio, Beijing, China). 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to separate the protein, which was subsequently moved to polyvinylidene fluoride (YA1701, Solarbio, Beijing, China). The membrane was blocked with 5% bovine serum albumin for 1 h, and the primary antibody was incubated at 4°C overnight. Afterward, the membrane was incubated with a secondary antibody (1:2000, ab205718), and protein bands were observed using a chemiluminescence apparatus (Image Quant LAS4000, GE Healthcare, Chicago, IL, USA) with ECL luminescent solution. The gray values of the protein bands were analyzed using Image J (v1.8.0.345, National Institutes of Health, Bethesda, MD, USA). GAPDH was used as an internal parameter to quantify protein expression levels.
The primary antibodies included the following: FABP4 (1:1000, ab92501), mTOR (1:1000, ab32028), phospho-mTOR (1:1000, ab137133), and GAPDH (1:1000, ab9485). All antibodies were purchased from Abcam, Cambridge, MA, USA.
Statistical analysis
GraphPad Prism 9.0 software (Inc., San Diego, CA, USA) was used to analyze and process the experimental data. Data are expressed as mean ± standard deviation. The t-test was employed to analyze the differences between the two groups. Variance analysis between groups was conducted using a one-way analysis of variance, and the significance of difference was analyzed using the Tukey multipole difference test. P < 0.05 was considered statistically significant.
RESULTS
LPS-induced myocardial dysfunction in H9c2 cells
In Figure 1a, FABP4 messenger ribonucleic acid (mRNA) expression was significantly increased in H9C2 cells following LPS treatment, indicating that FABP4 may be associated with myocardial dysfunction (P < 0.001). Figures 1b and c show the expression levels of IL-1β and TNF-α, determined by ELISA kit and qRT-PCR, respectively. At the RNA level, the mRNA expression of IL-1β and TNF-α was significantly higher after LPS treatment (P < 0.001). Similarly, at the molecular level, the LPS group exhibited considerably higher protein levels of TNF-α and IL-1β [Figure 1d and e] (P < 0.001).
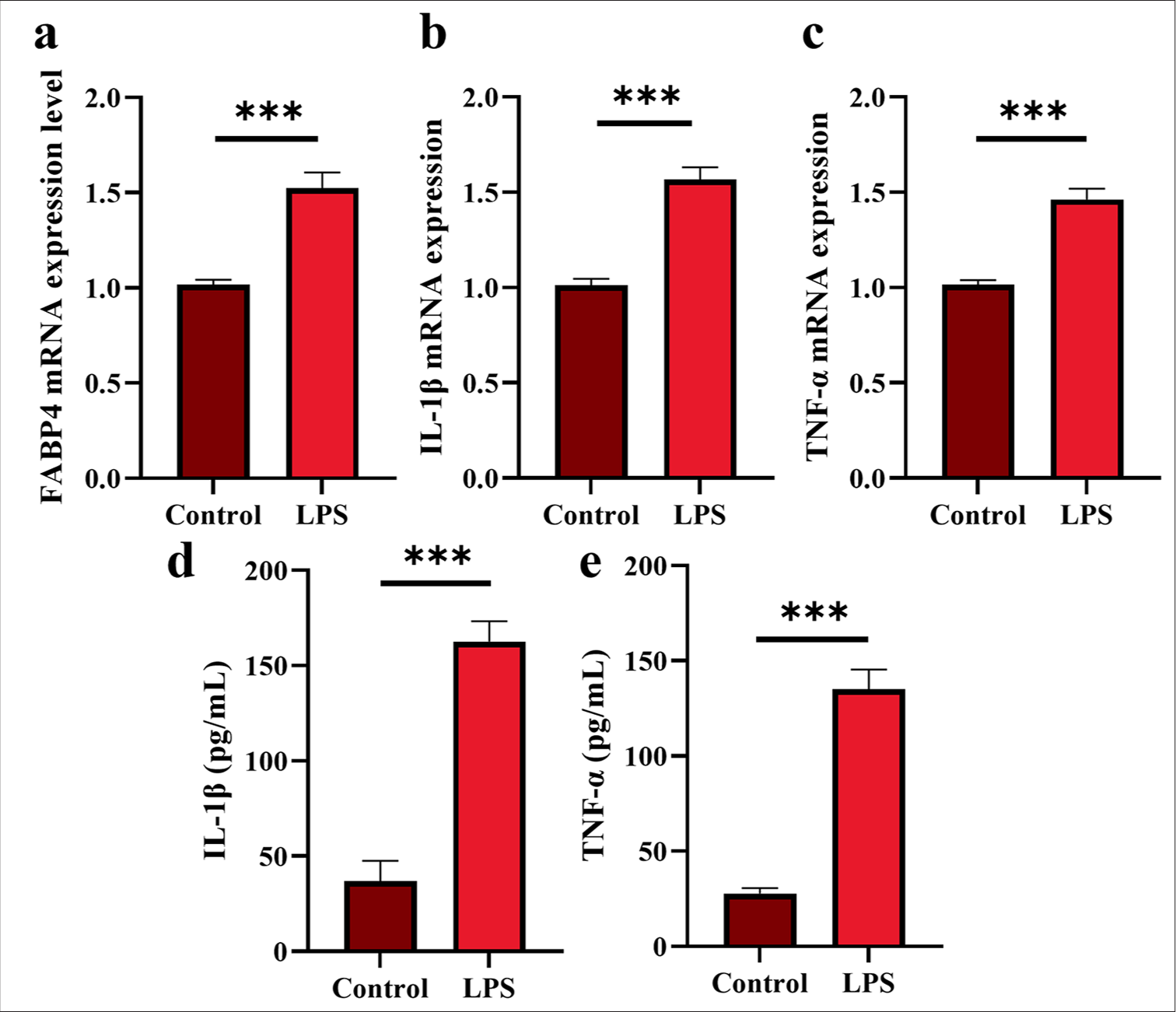
- LPS-induced myocardial dysfunction in H9c2 cells. (a) FABP4 mRNA expression. (b and c) IL-1β and TNF-α mRNA expression. (d and e) IL-1β and TNF-α expression. n = 3, ✶✶✶P < 0.001. LPS: Lipopolysaccharide, FABP4: Fatty acid-binding protein 4, TNF-α: Tumor necrosis factor-α, IL: Interleukin, mRNA: Messenger RibonucleicAcid.
Silencing FABP4 inhibits H9c2 cell inflammatory cytokines
Combined with the previous results, this study further explored the effect of FABP4 on SIMD by silencing its expression. Figures 2a and b show the levels of FABP4 in each group after different treatments. The expression of FABP4 was not statistically significant between the LPS and si-NC groups. However, in the si-FABP4 group, FABP4 expression was substantially reduced, confirming the successful silencing of FABP4. Subsequently, the secretion of proinflammatory factors in H9c2 cells was measured after FABP4 silencing [Figure 2c-f]. IL-1β and TNF-α expressions were considerably higher in the LPS group. At the mRNA and molecular levels, the IL-1β level was notably lower in the si-FABP4 group (P < 0.001). While the molecular level of TNF-α was significantly reduced (P < 0.001), the mRNA level was also significantly lower (P < 0.05) in the si-FABP4 group.
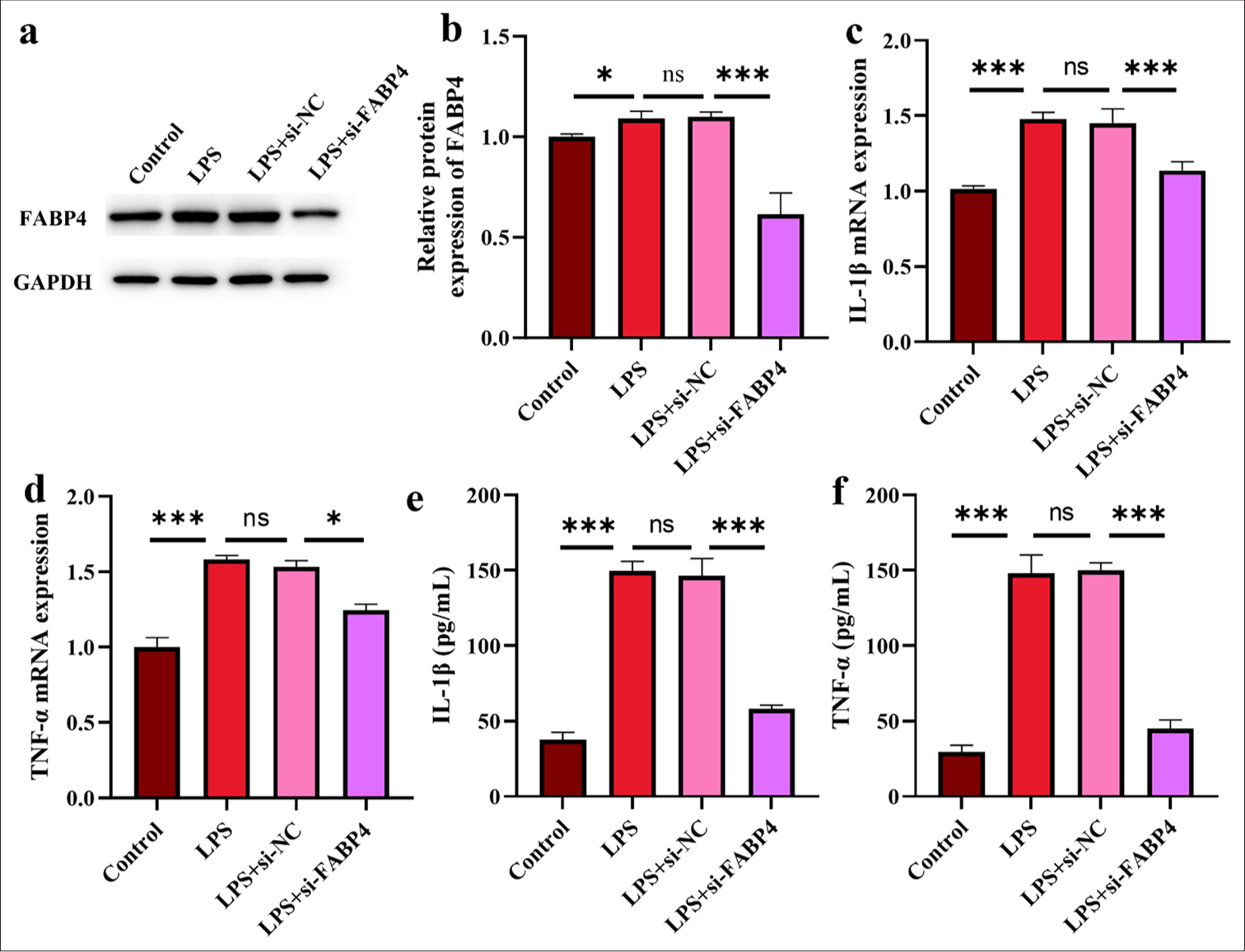
- Silencing FABP4 inhibits H9c2 cell inflammatory cytokines. (a and b) Silence FABP4 validation assay by Western blot. (c and d) IL-1β and TNF-α mRNA expressions. (e and f) IL-1β and TNF-α expressions. n = 3, ns: No statistical significance, ✶P < 0.05, ✶✶✶P < 0.001. FABP4: Fatty acid-binding protein 4, TNF-α: Tumor necrosis factor-α, IL: Interleukin, mRNA: Messenger RibonucleicAcid.
Silencing FABP4 inhibited H9c2 cell apoptosis
The experimental results of the CCK-8 assay, used to determine the viability of H9C2 cells, are shown in Figure 3a. After LPS treatment, H9c2 cell viability dramatically decreased (P < 0.001), whereas following FABP4 silencing, viability significantly increased (P < 0.05). Figure 3b shows that LDH values were significantly higher in the LPS and si-NC groups, but decreased significantly after FABP4 silencing (P < 0.001). In Figures 3c and d, the apoptotic cell rate in the LPS and si-NC groups increased significantly (P < 0.001), while that in the si-FABP4 group decreased significantly (P < 0.05). In Figure 3e-g, BAX-positive signal cells increased significantly in the LPS and si-NC groups, while the BAX-positive signal weakened, and the number of positive cells decreased significantly after FABP4 silencing (P < 0.001). The immunofluorescence staining pattern for Bcl-2 was the opposite of that for BAX. The rate of Bcl-2 positive cells decreased significantly in the LPS and si-NC groups, while the rate of Bcl-2 positive cells increased significantly after silencing FABP4 (P < 0.001). These results indicate that silencing FABP4 reduces LPS-induced apoptosis in myocardial dysfunction cells.
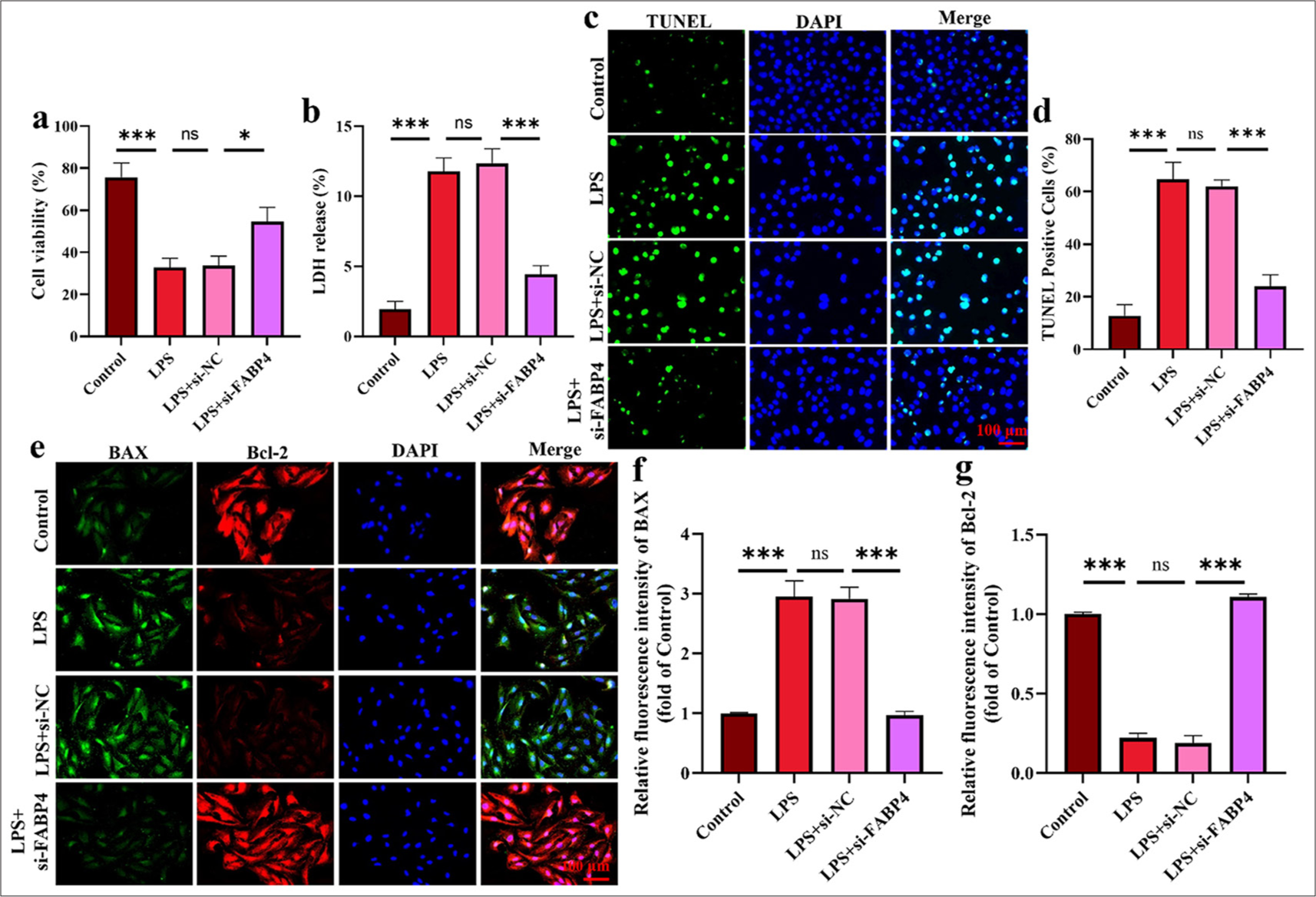
- Silencing FABP4 inhibited H9c2 cell apoptosis. (a) CCK-8 assay. (b) LDH ELISA assay. (c and d) TUNEL staining. (e-g) Immunofluorescence staining of BAX and Bal-2. n = 3, ns: No statistical significance, ✶P < 0.05, ✶✶✶P < 0.001. LDH: Layered double hydroxides, TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling, DAPI: 4’,6-diamidino-2-phenylindole, BAX: Bcl-2-associated X protein, Bcl-2: B-cell lymphoma-2, FABP4: Fatty acid-binding protein 4, CCK-8: Cell counting kit-8, ELISA: Enzyme-linked immunosorbent assay.
Silencing FABP4 increased the antioxidant activity of H9c2 cells
In Figures 4a and b, the ROS levels in the LPS group were noticeably higher (P < 0.001). After silencing FABP4, ROS levels decreased significantly (P < 0.001). Figure 4c and d show the results of JC-1 staining, indicating LPS-induced mitochondrial function in SIMD cells. After LPS treatment, the MMP in H9c2 cells was significantly reduced (P < 0.001), and this effect was significantly reversed by FABP4 silencing (P < 0.001). Subsequently, ATP levels were measured using an ELISA kit. Figure 4e shows that LPS significantly reduced ATP levels in H9c2 cells, while ATP levels significantly increased after FABP4 silencing (P < 0.01). Finally, iNOS and SOD2 were stained by immunofluorescence. The results shown in Figure 4f-i indicate that after LPS treatment, the rate of SOD2-positive cells in H9c2 cells significantly decreased, while the rate of iNOS-positive cells significantly increased (P < 0.001). However, after silencing FABP4, the trends in the rates of SOD2 and iNOS-positive cells were reversed. The rate of SOD2-positive cells in the si-FABP4 group was significantly upregulated, while that of iNOS-positive cells was significantly downregulated (P < 0.001). These data indicate that silencing FABP4 has a protective effect on LPS-induced H9c2 cells by alleviating mitochondrial dysfunction.
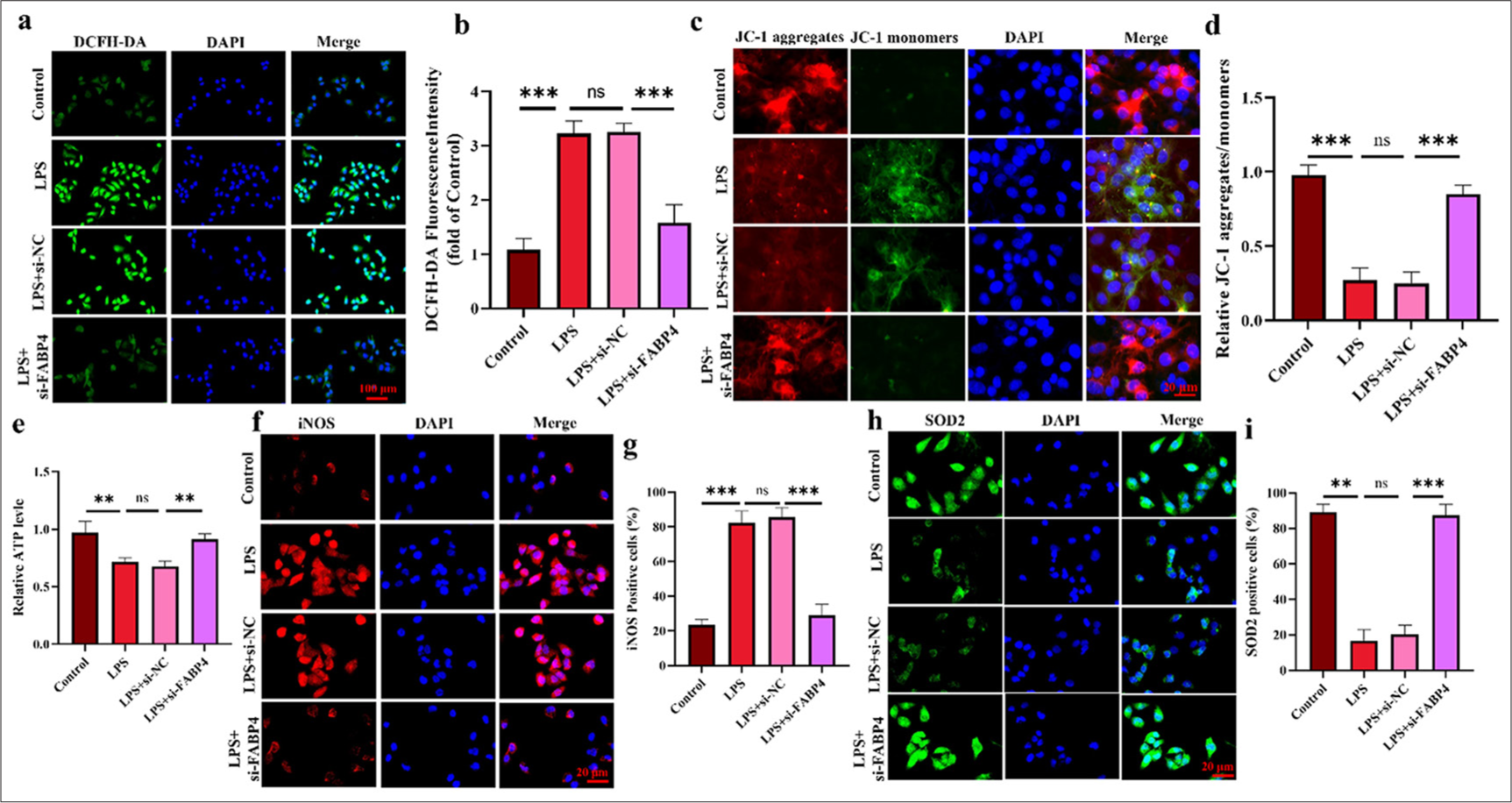
- Silencing FABP4 increased the antioxidant activity of H9c2 cells. (a and b) ROS levels were analyzed using DCFH-DA staining. (c and d) JC-1 staining assay. (e) ATP levels of H9c2 cells. (f-i) Immunofluorescence staining and quantification results of iNOS and SOD2. n = 3, ns: No statistical significance, ✶✶P < 0.01, ✶✶✶P < 0.001. DCFH-DA: 2’,7’-Dichlorodihydrofluorescein diacetate, JC-1: 5,5',6,6'-Tetrachloro-1,1',3,3'-tetraethylimidacarbocyanine, iNOS: Inducible nitric oxide synthase, SOD2: Superoxide dismutase 2, FABP4: Fatty acid-binding protein 4, ROS: Reactive oxygen species.
Silencing FABP4 inhibits H9c2 apoptosis through mTOR signaling
Figures 5a and b show that mTOR phosphorylation in H9c2 cells significantly decreased after LPS treatment. After FABP4 silencing, the phosphorylation level of mTOR increased significantly FABP4 silencing was combined with the mTOR signaling pathway inhibitor AZD-8055 to further demonstrate that PABF4 improves SIMD through this pathway. Figures 5c and d show that after silencing FABP4 in combination with AZD-8055, the phosphorylation level of mTOR decreased significantly. In Figure 5e and f, after combining with ZAD-8055, H9c2 cell viability decreased significantly (P < 0.001), and LDH level increased significantly (P < 0.01). Figure 5g and h showed that the anti-apoptotic capability of AZD-8055 cells was significantly reduced after treatment (P < 0.001). These results provide further evidence that FABP4 may ameliorate SIMD through the mTOR signaling pathway.
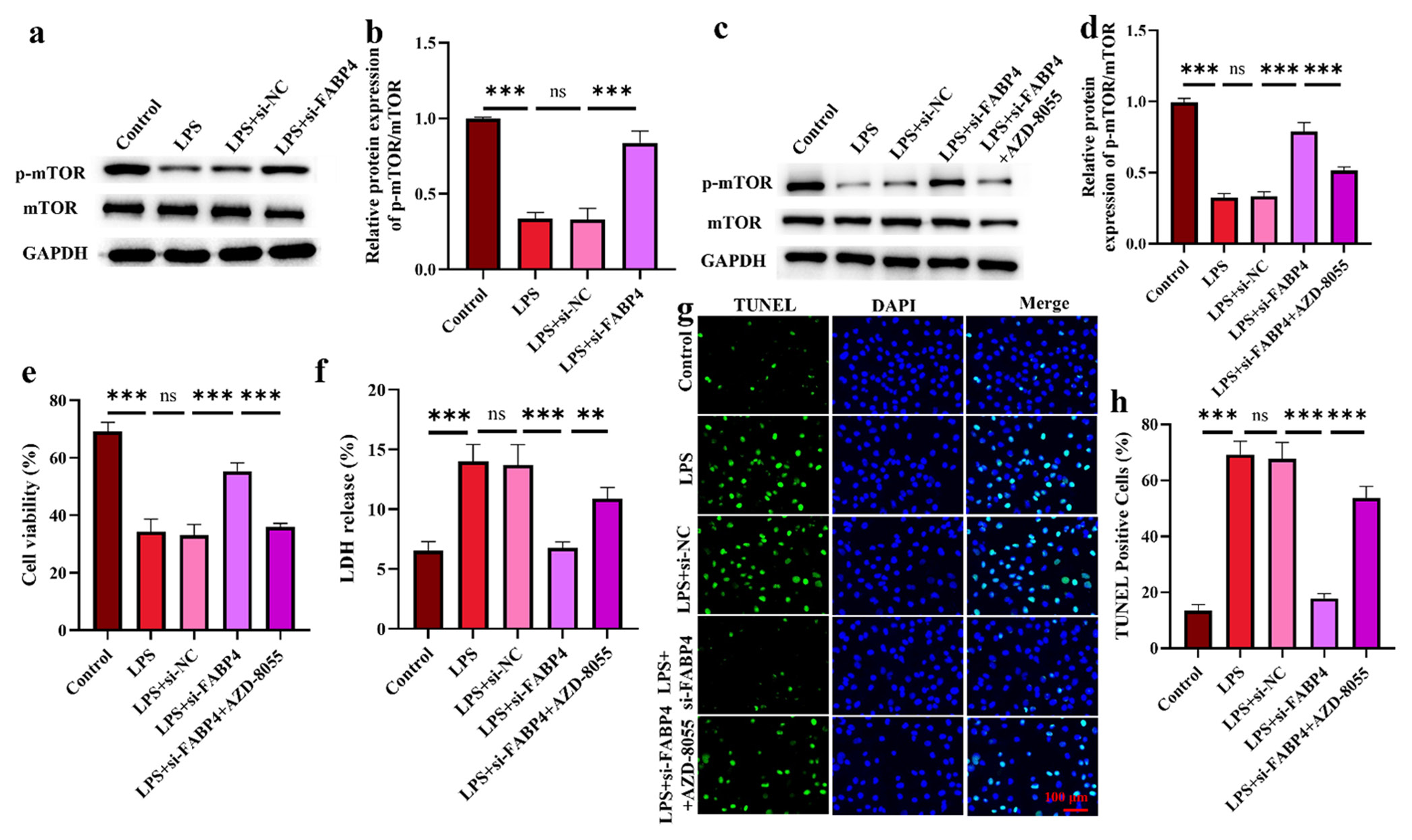
- Silencing FABP4 inhibits H9c2 apoptosis through mTOR signaling. (a-d) mTOR signaling pathway expression. (e) H9c2 cell viability. (f) LDH levels in H9c2 cells. (g and h) Apoptosis was determined by TUNEL staining. n = 3, ns: No statistical significance, ✶✶P < 0.01, ✶✶✶P < 0.001. mTOR: Mammalian target of rapamycin, FABP4: Fatty acid-binding protein 4, LDH: Layered double hydroxides, TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
Silencing FABP4 increases H9c2 antioxidant activity through mTOR signaling
Finally, the mitochondrial function of H9c2 cells was assessed to explore the influence of the mTOR signaling pathway on mitochondrial function. Figures 6a and b show that, after silencing FABP4 in combination with AZD-8055, ROS levels increased significantly (P < 0.001). The results of iNOS and SOD2 immunofluorescence staining showed that, after AZD-8055 treatment of H9c2 cells, iNOS-positive cells significantly increased, while SOD2-positive cells significantly decreased [Figure 6c-f] (P < 0.001). Finally, ATP levels in H9c2 cells treated with AZD-8055 were measured. Figure 6g shows that ATP levels in H9c2 cells decreased significantly after AZD-8055 intervention (P < 0.05). These results indicate that the downstream molecular mechanism of FABP4 may involve the mTOR signaling pathway.
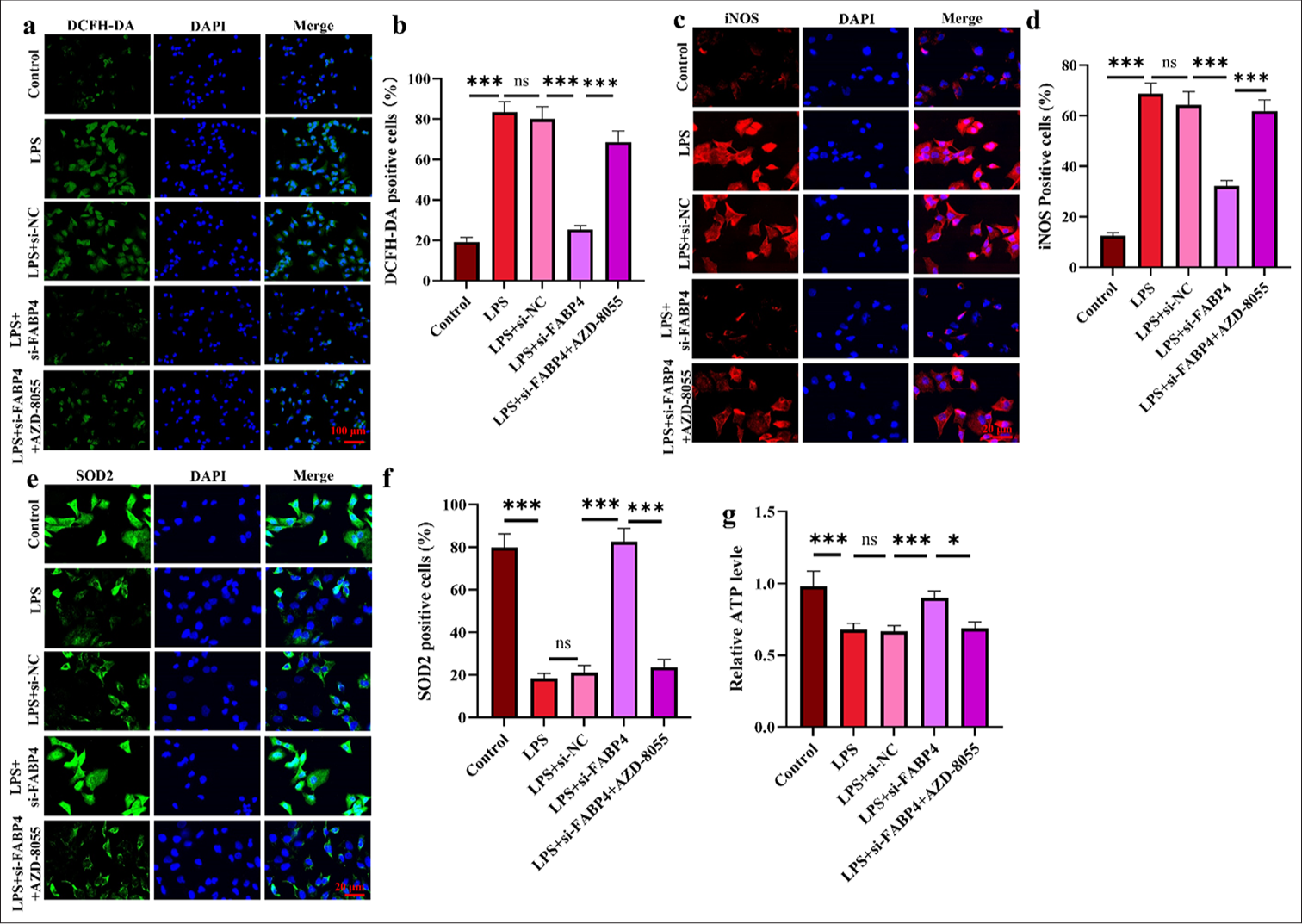
- Silencing FABP4 increases H9c2 antioxidant activity through mTOR signaling. (a and b) ROS levels were analyzed through DCHF–DA staining. (c-f) Immunofluorescence staining and quantification results of iNOS and SOD2. (g) ATP level in H9c2 cells. n = 3, ns: No statistical significance, ✶P < 0.05, ✶✶✶P < 0.001. FABP4: Fatty acid-binding protein 4, ROS: Reactive oxygen species, mTOR: Mammalian target of rapamycin, DCFH-DA: 2’,7’-Dichlorodihydrofluorescein diacetate, iNOS: Inducible nitric oxide synthase, SOD2: Superoxide dismutase 2, ATP: Adenosine triphosphate.
DISCUSSION
The pathophysiological mechanism of SIMD can be divided into intracellular and extracellular mechanisms.[4] The intracellular mechanisms mainly include calcium regulation disorders, mitochondrial dysfunction, autonomic nervous system disorders, and cardiac autophagy. Meanwhile, extracellular mechanisms include circulatory and microvascular changes, skin dysfunction, and myocardial inhibitory factors.[1,21] The current research focuses on the transfer of SIMD to the pathophysiological mechanism through mitochondrial function signals. During septic cardiomyopathy, mitochondrial dysfunction leads to impaired mitochondrial function and increased mitochondrial division, leading to the excessive release of ROS, protein, and lipid molecules. Oxygen free radicals rapidly damage DNA, eventually leading to cell death.[22,23] The primary source of intracellular ROS is the mitochondria, and studies have shown for many years that LPS promotes mitochondrial ROS formation and oxidative stress in the heart, which can lead to mitochondrial damage and cardiac dysfunction.[24,25] The body’s reaction to LPS plays a major role in the myocardial cell death associated with sepsis. After blocking this response, cell viability increased, and mitochondrial damage was alleviated.[26-28] Thus, LPS was used to induce the SIMD cell model, and the results showed a considerable increase in the expressions of TNF-α and IL-1β, a reduction in the anti-apoptosis capability of cells, and impaired mitochondrial function, indicating that the SIMD model was successfully constructed. In addition, mitochondria are considered signaling organelles. They have been found to regulate various forms of cell death (apoptosis, pyrodeath, and necrosis).[29,30] By controlling the outer membrane’s permeability, the Bcl-2 family enables mitochondria to regulate cell death.[29] This phenomena is also supported by the study’s findings. Immunofluorescence labeling for Bcl-2 and BAX was used to demonstrate the anti-apoptotic potential of H9c2 cells. The findings showed that LPS decreased H9C2 cells’ resistance to apoptosis.
The link between FABP4 and metabolic and inflammatory pathways in adipose tissue (fat cells and macrophages) contributes to the development of metabolic disorders and cardiovascular diseases.[31-33] This study indicates that small-molecule inhibition of FABP4 could be a promising therapeutic strategy for insulin resistance and atherosclerosis.[34] First, FABP4 expression in H9c2 cells was examined, revealing that normal rat cardiomyocytes had high FABP4 expression. Therefore, by suppressing FABP4 expression, it was shown that FABP4 protects against LPS-induced SIMD. The results showed that silencing FABP4 exerted a protective effect against SIMD by enhancing the anti-apoptotic capability, increasing cell viability, and improving mitochondrial dysfunction in H9c2 cells. mTOR phosphorylation was measured, and the results illustrated that silencing FABP4 improves SIMD by promoting phosphorylation of the mTOR signaling pathway. FABP4 silencing was also combined with mTOR inhibitor AZD-8055, further verifying that mTOR is a key target pathway of SIMD. The outcomes revealed that AZD-8055 therapy reduced the capacity of H9c2 cells to prevent apoptosis, which inhibited the recovery of mitochondrial dysfunction and the enhancement of cell vitality. In addition, FABP4 may induce chondrocyte degeneration through nuclear factor-kappa B signaling[35] and promote adipogenesis in human skeletal muscle cells by mediating the peroxisome proliferator-activated receptor γ pathway.[36] These results provide a direction for further research on other action targets of FABP4.
This study still has several limitations. First, only rat cardiomyocyte H9c2 was used in this study. Thus, future studies should use animal models and patient-derived samples to further prove the assumption. In addition, only the phosphorylation level of mTOR was discussed. Therefore, other signaling pathways should be identified to enrich molecular events for the treatment of SIMD.
SUMMARY
H9c2 cells were treated with LPS in this study to establish the SIMD cell model. The results revealed that silencing FABP4 increased cell viability, inhibited LDH levels, and enhanced the anti-apoptotic capability and mitochondrial function of cells. Furthermore, silencing FABP4 improves SIMD by promoting mTOR phosphorylation.
ACKNOWLEDGMENT
Not applicable.
AVAILABILITY OF DATA AND MATERIAL
The datasets used and/or analyzed during the present study were available from the corresponding author on reasonable request.
ABBREVIATIONS
CCK-8: Cell counting kit-8
DAPI: 4’,6-diamidino-2-phenylindole
DMEM: Dulbecco’s modified eagle medium
ELISA: Enzyme-linked immunosorbent assay
FABP4: Fatty acid-binding protein 4
LPS: Lipopolysaccharide
MMP: Mitochondrial membrane potential
PVDF: Polyvinylidene fluoride
RIPA: Radio immunoprecipitation assay
SDS-PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis
SIMD: Sepsis-induced myocardial dysfunction
TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
AUTHOR CONTRIBUTIONS
ZLQ and KXZ: Designed the study; all authors conducted the study; ZLQ and WC: Collected and analyzed the data; QYQ and WC: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, take public responsibility for appropriate portions of the content, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work are appropriately investigated and resolved.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE:
Ethical approval and consent to participate is not required as this study does not involve animal or human experiments.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
EDITORIAL/PEER REVIEW
To ensure the integrity and highest quality of CytoJournal publications, the review process of this manuscript was conducted under a double-blind model (authors are blinded for reviewers and vice versa) through an automatic online system.
FUNDING
Not applicable.
References
- Sepsis-induced cardiac dysfunction: A review of pathophysiology. Acute Crit Care. 2020;35:57-66.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiovascular clusters in septic shock combining clinical and echocardiographic parameters: A post hoc analysis. Intensive Care Med. 2019;45:657-67.
- [CrossRef] [PubMed] [Google Scholar]
- Sepsis-induced myocardial dysfunction (SIMD): The pathophysiological mechanisms and therapeutic strategies targeting mitochondria. Inflammation. 2020;43:1184-200.
- [CrossRef] [PubMed] [Google Scholar]
- Correction to: Immunometabolism: Another road to sepsis and its therapeutic targeting. Inflammation. 2019;42:789.
- [CrossRef] [PubMed] [Google Scholar]
- Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776-87.
- [CrossRef] [PubMed] [Google Scholar]
- Bench-to-bedside review: Endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;10:233.
- [CrossRef] [PubMed] [Google Scholar]
- MyD88-dependent expansion of an immature GR-1(+)CD11b (+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463-74.
- [CrossRef] [PubMed] [Google Scholar]
- Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594-605.
- [CrossRef] [PubMed] [Google Scholar]
- Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med. 2013;41:820-32.
- [CrossRef] [PubMed] [Google Scholar]
- Importance of fatty acid binding proteins in cellular function and organismal metabolism. J Cell Mol Med. 2024;28:e17703.
- [CrossRef] [PubMed] [Google Scholar]
- Proteomic profiling of fatty acid binding proteins in muscular dystrophy. Expert Rev Proteomics. 2020;17:137-48.
- [CrossRef] [PubMed] [Google Scholar]
- Adipocyte fatty acid-binding protein as a determinant of insulin sensitivity in morbid-obese women. Obesity (Silver Spring). 2009;17:1124-8.
- [CrossRef] [PubMed] [Google Scholar]
- Reduction of serum FABP4 level by sitagliptin, a DPP-4 inhibitor, in patients with type 2 diabetes mellitus. J Lipid Res. 2015;56:2372-80.
- [CrossRef] [PubMed] [Google Scholar]
- Predictive and diagnostic biomarkers for gestational diabetes and its associated metabolic and cardiovascular diseases. Cardiovasc Diabetol. 2019;18:140.
- [CrossRef] [PubMed] [Google Scholar]
- Relationships between visceral/subcutaneous adipose tissue FABP4 expression and coronary atherosclerosis in patients with metabolic syndrome. Cardiovasc Pathol. 2020;46:107192.
- [CrossRef] [PubMed] [Google Scholar]
- Adipocyte fatty acid binding protein 4 (FABP4) inhibitors An update from 2017 to early 2022. Eur J Med Chem. 2022;240:114604.
- [CrossRef] [PubMed] [Google Scholar]
- Critical role of mTOR, PPARγ and PPARδ signaling in regulating early pregnancy decidual function, embryo viability and feto-placental growth. Mol Hum Reprod. 2018;24:327-40.
- [CrossRef] [PubMed] [Google Scholar]
- Role of the mTOR signalling pathway in human sepsis-induced myocardial dysfunction. Can J Cardiol. 2019;35:875-83.
- [CrossRef] [PubMed] [Google Scholar]
- Gracillin inhibits apoptosis and inflammation induced by lipopolysaccharide (LPS) to alleviate cardiac injury in mice via improving miR-29a. Biochem Biophys Res Commun. 2020;523:580-7.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac function and dysfunction in sepsis. Clin Chest Med. 2016;37:289-98.
- [CrossRef] [PubMed] [Google Scholar]
- Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 2019;130:160-9.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017;2:94248.
- [CrossRef] [PubMed] [Google Scholar]
- Polyethylene glycol-superoxide dismutase prevents endotoxin-induced cardiac dysfunction. Am J Respir Crit Care Med. 2006;173:1240-7.
- [CrossRef] [PubMed] [Google Scholar]
- Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem. 2003;278:41510-8.
- [CrossRef] [PubMed] [Google Scholar]
- Intracellular lipopolysaccharide sensing as a potential therapeutic target for sepsis. Trends Pharmacol Sci. 2019;40:187-97.
- [CrossRef] [PubMed] [Google Scholar]
- lncRNA RMRP prevents mitochondrial dysfunction and cardiomyocyte apoptosis via the miR-1-5p/hsp70 Axis in LPS-induced sepsis mice. Inflammation. 2020;43:605-18.
- [CrossRef] [PubMed] [Google Scholar]
- Shenfu injection prevents sepsis-induced myocardial injury by inhibiting mitochondrial apoptosis. J Ethnopharmacol. 2020;261:113068.
- [CrossRef] [PubMed] [Google Scholar]
- The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426-31.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21:193-201.
- [CrossRef] [PubMed] [Google Scholar]
- Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377-9.
- [CrossRef] [PubMed] [Google Scholar]
- Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699-705.
- [CrossRef] [PubMed] [Google Scholar]
- Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640-50.
- [CrossRef] [PubMed] [Google Scholar]
- Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959-65.
- [CrossRef] [PubMed] [Google Scholar]
- Fatty acid binding protein 4 (FABP4) induces chondrocyte degeneration via activation of the NF-κb signaling pathway. FASEB J. 2024;38:e23347.
- [CrossRef] [PubMed] [Google Scholar]
- Interleukin-4-induced FABP4 promotes lipogenesis in human skeletal muscle cells by activating the PPAR γ signaling pathway. Cell Biochem Biophys. 2022;80:355-66.
- [CrossRef] [PubMed] [Google Scholar]




