


Translate this page into:
Serous fluid: Metastatic sarcomas, melanoma, and other non-epithelial neoplasms

-
Received: ,
Accepted: ,
How to cite this article: Pantanowitz L, Chivukula M. Serous fluid: Metastatic sarcomas, melanoma, and other non-epithelial neoplasms. CytoJournal 2022;19:15.
HTML of this article is available FREE at: https://dx.doi.org/10.25259/CMAS_02_10_2021
Abstract
While most tumors metastatic to the serous membranes are of epithelial origin, cytologists should be aware that non-epithelial neoplasms can also cause malignant effusions including sarcomas, melanomas, germ cell tumors, and, more rarely, brain tumors. The differential diagnosis of a malignant effusion is accordingly broad, especially for the small round blue cell tumors that includes not only mesenchymal tumors, but also non-mesenchymal tumors, such as neuroblastoma and Wilms tumor. Diagnosing non-epithelial malignancies in effusion specimens based entirely upon their cytomorphologic features is difficult because these neoplasms often exhibit considerable morphological overlap and their cytomorphology can differ from the original tumor. As malignant cells have a tendency to round up in body fluids these non-epithelial neoplasms can therefore mimic reactive mesothelial cells and metastatic adenocarcinoma. The use of ancillary studies including immunostaining, FISH, and molecular studies is thus often critical to reach a definitive diagnosis. This review article will be incorporated finally as one of the chapters in CMAS (CytoJournal Monograph/Atlas Series) #2. It is modified slightly from the chapter by the initial authors in the first edition of Diagnostic Cytopathology of Serous Fluids.
Keywords
Effusion
germ cell tumor
malignant
melanoma
metastasis
sarcoma
serous fluid


INTRODUCTION
Most tumors metastatic to the serous membranes are of epithelial origin. Sarcomas account for only 3–6% of malignant effusions and usually occur in the setting of a known primary tumor. Common non-epithelial neoplasms that may cause malignant effusions include malignant melanoma, sarcomas, and other neoplasms including germ cell tumors [Figure 1]. Other rare non-epithelial malignancies that may be encountered in effusion cytology include neuroblastoma, Wilms tumor, and metastatic brain tumors.1-3 Intracranial brain tumors, most commonly medulloblastoma [Figure 2], can metastasize to the peritoneal cavity via ventriculo-peritoneal shunts used to divert excess cerebrospinal fluid.4 Malignant effusions caused by non-epithelial neoplasms are more frequently encountered in children than in adults. However, the most common causes of malignant effusions in children are lymphoma and leukemia, followed by non-epithelial neoplasms including Wilms tumor, neuroblastoma, Ewing sarcoma, and embryonal rhabdomyosarcoma [Figure 1].5
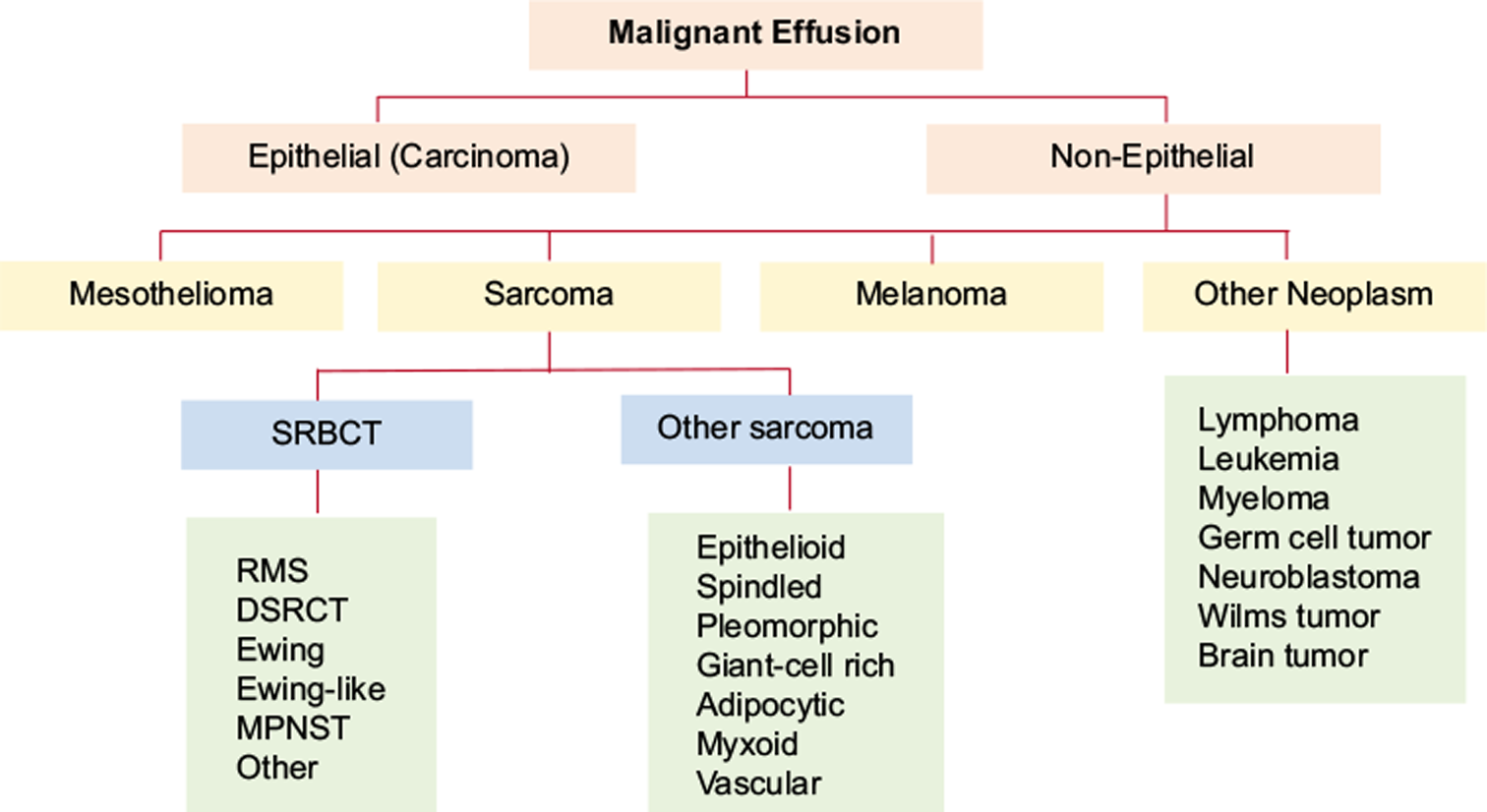
- Algorithm summarizing a diagnostic approach to non-epithelial causes of malignant effusion.
- DSRCT = desmoplastic small round cell tumor, MPNST = malignant peripheral nerve sheath tumor, RMS = rhabdomyosarcoma, SRBCT = small round blue cell tumor
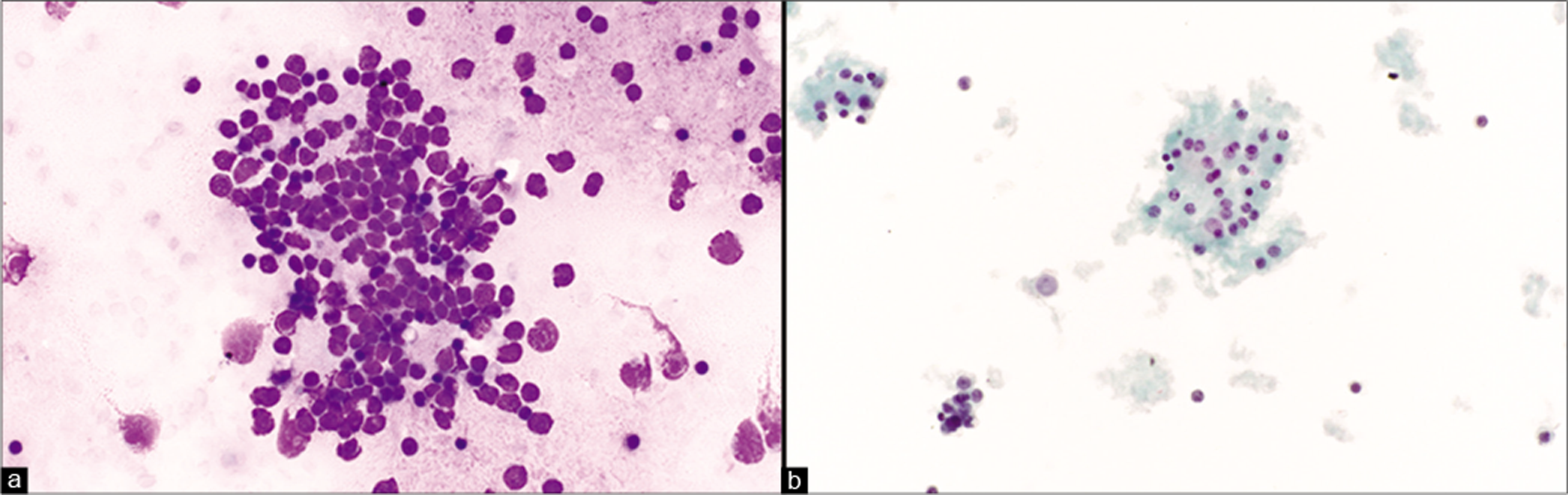
-
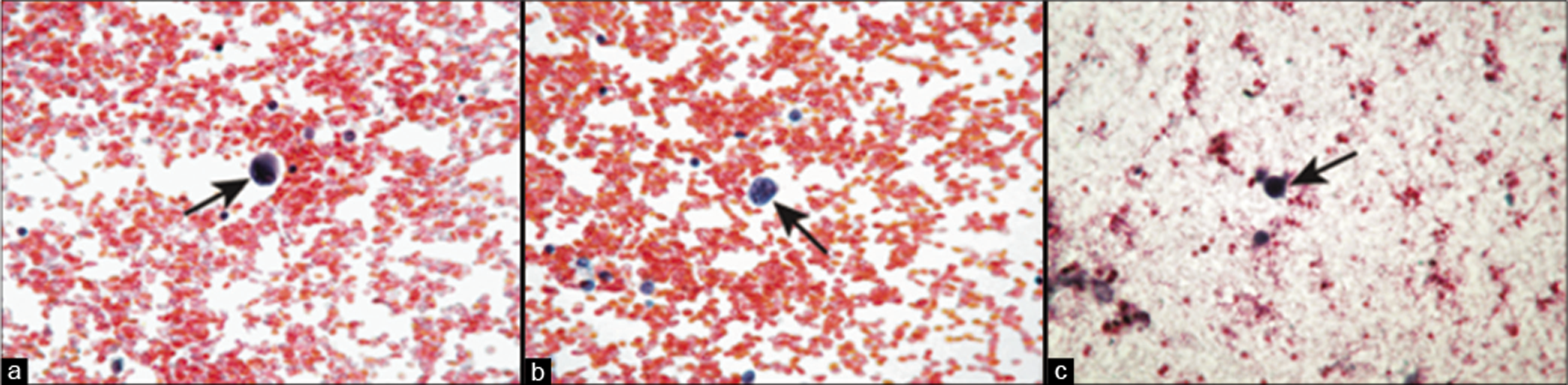
Medulloblastoma metastatic to pleural fluid. Tumor cells are characterized as small round blue cells (a). On ThinPrep the small tumor cells can be easily mistaken for chronic inflammatory cells (b). [a. Smear, DQ stain, 40x; b. ThinPrep, Pap stain, 40x]
Determining the origin of non-epithelial malignancies that are metastatic to body fluids based solely upon their cytomorphologic features is difficult. The reason is that these tumors often have considerable overlapping morphologic features and their cytomorphology can differ from those of the original tumor. For example, spindle cell tumors and even small round blue cell tumors (SRBCTs) often show epithelioid cells that can present as single isolated cells or even in large, cohesive clusters mimicking metastatic carcinoma.6 The diagnosis thus often requires correlation with clinical history, comparative review of the metastatic tumor cells in the effusions with the original tumor cells, and the use of ancillary studies.7
GENERAL CYTOLOGY
Several studies have described the cytologic examination of exfoliated sarcomas in effusion fluids.6-8 In general, due to the surface tension from the surrounding fluid, sarcoma cells have a tendency to round up even if they were spindle shaped in the original tumor [Figure 3]. Moreover, metastases in effusions usually lack helpful tissue arrangement because of which the tumor cell growth, vasculature, or stromal patterns can not be observed. Metastatic sarcomas in fluids can be variably cellular and are arranged either as single tumor cells or in small clusters. Sarcomatous cells predominantly assume a polyhedral form with varying amounts of cytoplasm and indistinct borders, but may show bipolar cytoplasmic processes. Binucleation or multinucleation may be present in certain non-epithelial malignancies (e.g. alveolar rhabdomyosarcoma, melanoma). The nuclei of tumor cells are usually round to oval, and sometimes even fusiform or spindle-shaped. Nuclei of malignant cells usually do not demonstrate marked irregular contours, but may reveal some nuclear membrane infolding along with chromatin clumping and prominent nucleoli. The nuclear details are appreciated better in ThinPrep preparations due to the monolayer arrangement of cells obtained with this method.9 Mitotic figures may be observed in any high-grade malignancy. Sarcomas can be accompanied by a tumor diathesis including a proteinaceous background with lysed blood and inflammatory cells.10 ThinPrep preparations tend to have a cleaner background and may lack such a tumor diathesis. A myxoid matrix associated with some sarcomas is better appreciated in cell-blocks,11-14 which can be highlighted by Alcian blue staining at pH 2.5.15
- Pelvic washings positive for metastatic uterine malignancy. This uterine leiomyosarcoma shows numerous pleomorphic epithelioid tumor cells and a rare multinucleated cell present in a background of inflammatory cells with blood (a). This malignant mixed müllerian tumor (carcinosarcoma) contains both clusters of carcinoma cells and isolated sarcoma cells (b). [a. Cytospin, Pap stain, 20x; b. Cytospin, Pap stain, 20x]
ANCILLARY STUDIES
Ancillary studies such as immunohistochemistry (IHC) with SCIP (Subtractive Coordinate Immunoreactivity Pattern) approach,16 fluorescence in situ hybridization (FISH), molecular testing, and cytogenetics17 can be employed for confirmation and characterization of many metastatic malignancies in serous effusions, including sarcomas.18 While sarcomas such as synovial sarcoma and epithelioid sarcoma usually express keratins, it is important to be aware that several other sarcomas (e.g. angiosarcoma, osteosarcoma) may aberrantly express epithelial immunomarkers such as cytokeratin and epithelial membrane antigen (EMA).
Melanocytic immunomarkers including SOX10, HMB45, Melan-A /Mart-1, tyrosinase, MITF, and PRAME are useful in confirming melanoma. S-100 protein can be expressed not only in melanoma, but also in liposarcoma, chondrosarcoma, sarcomas with neural differentiation, and myoepithelial tumors. Muscle immunomarkers such as actin, desmin, myoglobin, myogenin, and h-caldesmon are helpful in identifying sarcomas with myogenic differentiation. Nuclear positivity for MyoD1 is observed in tumors with striated muscle differentiation (rhabdomyosarcoma) with high sensitivity. Several newer immunomarkers have emerged that are specific for certain soft tissue tumors such as CAMTA1 (epithelioid hemangioendothelioma), DOG1 (gastrointestinal stromal tumor), STAT6 (solitary fibrous tumor) and SS18-SSX (synovial sarcoma). High-grade undifferentiated sarcomas, that have a high propensity to metastasize, may be difficult to workup because they often do not exhibit a specific immunophenotype. FISH testing is also available to aid in the diagnosis of soft tissue tumors such as EWSR1 (Ewing sarcoma, desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma), MDM2 (liposarcoma), DDIT3 (myxoid/ round cell liposarcoma), SS18 (synovial sarcoma), and FOXO1 (alveolar rhabdomyosarcoma).
SARCOMAS
Small round blue cell tumors (SRBCTs)
SRBCTs are a subgroup of tumors [Table 1] that are usually cytologically indistinguishable and may thus create a diagnostic dilemma [Table 2].19,20 The use of ancillary studies is often critical to reach a definitive diagnosis [Table 3]. The differential diagnosis includes not only mesenchymal tumors, but also lymphoma/leukemia, small cell carcinoma, Merkel cell carcinoma, neuroblastoma, and melanoma with small cell morphology. Differentiated soft tissue tumors with small cell features include rhabdomyosarcoma, desmoplastic small round cell tumor (DSRCT), high-grade (round cell) myxoid liposarcoma, poorly differentiated synovial sarcoma (small cell variant), extraskeletal mesenchymal chondrosarcoma, small cell osteosarcoma, and malignant peripheral nerve sheath tumor (MPNST) with small cell pattern. Undifferentiated small round cell sarcomas include Ewing sarcoma, round cell sarcoma with EWSR1-non-ETS fusions, CIC-rearranged sarcoma, and sarcoma with BCOR genetic alterations. Many of these newer undifferentiated small round cell sarcomas were previously diagnosed as atypical Ewing sarcoma (or Ewing-like), because while they were morphologically similar to Ewing sarcoma they lacked typical immunophenotypic or molecular features.
| Mesenchymal Tumors | Non-Mesenchymal Tumors | |
|---|---|---|
| Differentiated soft tissue tumors | Undifferentiated sarcomas | |
| Rhabdomyosarcoma Desmoplastic small round cell tumor Round cell/high-grade myxoid liposarcoma Mesenchymal chondrosarcoma Poorly differentiated synovial sarcoma (small cell variant) Small cell osteosarcoma Malignant peripheral nerve sheath tumor with small cell change Melanotic neuroectodermal tumor of infancy |
Ewing sarcoma Round cell sarcoma with EWSR1-non-ETS fusions CIC-rearranged sarcoma BCOR-rearranged sarcoma |
Lymphoma/leukemia Neuroblastoma Small cell (neuroendocrine) carcinoma Merkel cell carcinoma Small cell melanoma Wilms tumor |
| Small blue cell tumor | Features | Lymphoma | Small cell carcinoma | RMS | Ewing sarcoma | DSRCT | Wilms tumor | Neuroblastoma |
|---|---|---|---|---|---|---|---|---|
| Cellular pattern | Single dyshesive cells | Linear chains, onion-skin clusters | Singly scattered or loose clusters Binucleation or multinucleation | Loose groups or single cells | Small clusters | Clusters or single cells | Clusters, single cells or rosettes | |
| Cytoplasm | Scant to abundant cytoplasm | Scant and delicate | Scant to abundant Cross striations indistinct | Scant ill-defined cytoplasm Intracytoplasmic glycogen | Scant, delicate with vacuoles | Minimal cytoplasm | Minimal cytoplasm | |
| Nucleus | Shape | Small, ovoid | Round | Small, ovoid | Small, ovoid | Small, ovoid, | Small, ovoid | Small, ovoid, fusiform, irregularly shaped |
| Chromatin | Clumped | Finely granular | Dense | Finely granular | Finely granular | Dense | Coarsely clumped | |
| Nucleoli | Often absent | Inconspicuous | Absent | Often prominent | Small and multiple | Absent | Inconspicuous | |
DSRCT: desmoplastic small round cell tumor; RMS: rhabdomyosarcoma
| Neoplasm | Keratin | S100 | LCA | CD99 | Desmin | Myogenin | WT1 | Other stains |
|---|---|---|---|---|---|---|---|---|
| Small cell carcinoma | + | - | - | - | - | - | - | Synaptophysin, Chromogranin, CD56, INSM1 |
| Lymphoblastic lymphoma | - | - | + | - | - | - | - | CD20, PAX5, CD43, TdT |
| Melanoma | - | + | - | - | - | - | - | SOX10, Melan-A/MART1, HMB-45, MiTF, Tyrosinase, PRAME |
| Alveolar RMS | + | - | - | + | + | + | + | Actin, MyoD1, ALK1, PAX5, Synaptophysin, CD56 |
| Embryonal RMS | + | + | - | - | + | + | - | Actin, MyoD1 |
| Ewing sarcoma | + | + | - | + | - | - | - | NKX2-2, ERG, Synaptophysin, Chromogranin, CD56 |
| CIC sarcoma | + | - | - | + | - | - | + | ETV4 |
| BCOR sarcoma | - | - | - | + | - | - | - | BCOR |
| DSRCT | + | - | - | + | + | - | + | EMA, NSE, GFAP, ERG, Synaptophysin, AR, CD117 |
| Synovial sarcoma | + | + | - | + | - | - | - | SS18-SSX, EMA, CD56, Calretinin |
| Neuroblastoma | - | + | - | - | - | - | - | PHOX2B, Synaptophysin, Chromogranin, NSE, PGP 9.5, INSM1, SOX10, CEA, CD117 |
| Wilms tumor | + | + | - | + | + | + | + | PAX8, Myoglobin, NFP, NSE, CD56, Glypican-3, GFAP, INI1, SALL4, CD117, CD31 |
| Mesenchymal chondosarcoma | - | + | - | + | + | - | + | SOX9, MYOD1, NSE, NKX2-2, SATB2 |
| Round cell liposarcoma | - | + | - | - | - | - | - | DDIT3, PRAME |
DSRCT: desmoplastic small round cell tumor; RMS: rhabdomyosarcoma
Rhabdomyosarcoma
Rhabdomyosarcoma is the most common tumor of adolescents and young adults and is rare in patients older than 40 years of age. There are four major histological subtypes: embryonal (most common), alveolar, sclerosing/ spindle cell, and pleomorphic. The most common sites of metastasis are the lungs, heart, and lymph nodes. Involvement of serous membranes as well as intraperitoneal spread can occur. Rhabdomyosarcomas can also occur in the setting of a sarcomatous transformation in patients with germ cell tumors or malignant mixed Müllerian tumors (MMMT) of the ovary or endometrium.21 The presence of a moderate or massive effusion is an unfavorable prognostic factor in children with rhabdomyosarcoma.22 Cytologic material from pleural, peritoneal, and pericardial fluids may be diagnostic of metastasis [Figure 4], but usually require the help of ancillary studies.23-25 By immunohistochemistry they show nuclear immunoreactivity for myogenin and MyoD1 with cytoplasmic immunoreactivity for desmin, as well as myoglobin in more differentiated cells. These stains may only be focal in myogenic tumor cells.
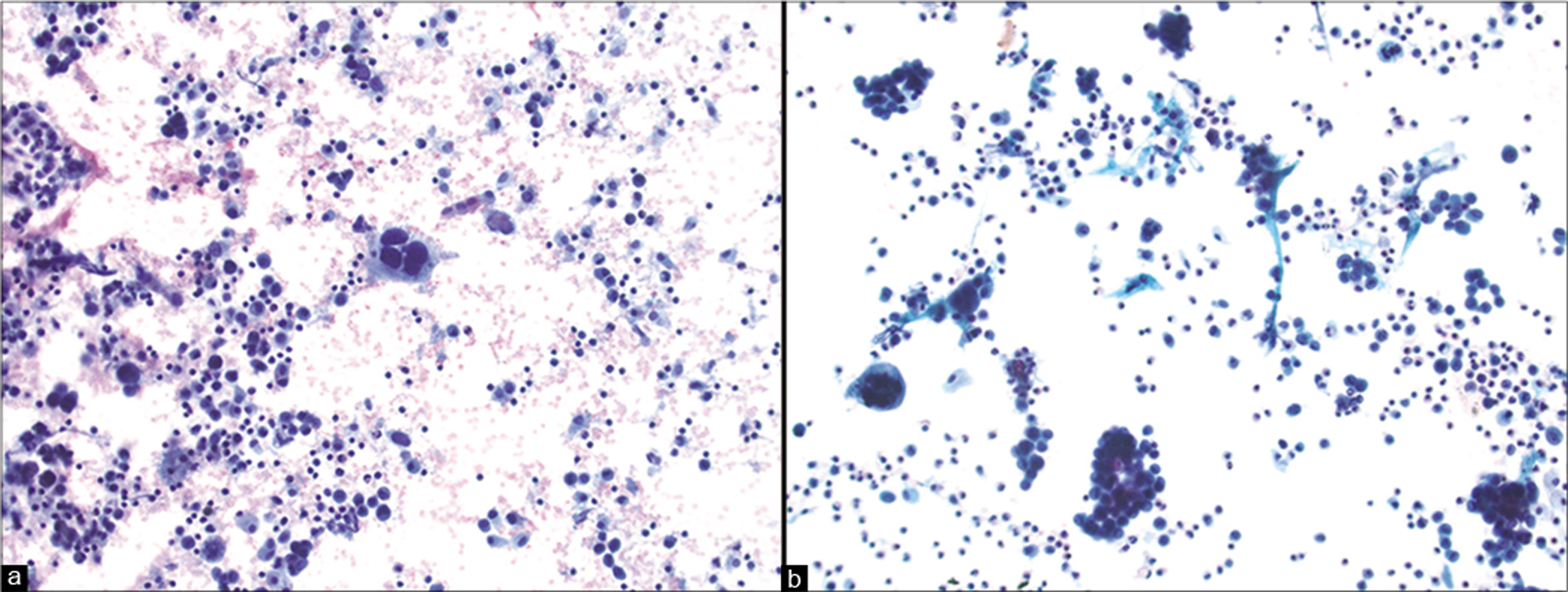
- Pleural fluid samples from different patients with metastatic rhabdomyosarcoma. Tumor cells are typically characterized as small round cells with minimal cytoplasm (a). Tumor cells can sometimes have more moderate cytoplasm and nuclear pleomorphism (b). Tumor cells can form clusters (c). Multinucleated giant cells as shown in the center of the cell-block may be seen with alveolar rhabdomyosarcoma (d). [a. Smear, Pap stain, 60x; b. Smear, DQ stain, 40x; c. Smear, DQ stain, 40x; d. cell-block, H&E stain, 40x]
Embryonal rhabdomyosarcoma accounts for nearly 80% of all rhabdomyosarcomas. The most common site of occurrence is the head and neck, followed by genitourinary (e.g. sarcoma botryoides) and paratesticular locations, as well as the extremities. Metastases to the body fluids have been reported.26 This sarcoma shows aggregates of small cells, which are about 2–5 times the size of mature lymphocytes. Malignant samples may also contain scattered large isolated cells with conspicuous perinuclear clearing. Their nuclei are typically enlarged, hyperchromatic, and pleomorphic. The cytoplasm of tumor cells is somewhat bubbly with indistinct borders. In Diff-Quik (DQ)-stained smears, the cells appear poorly differentiated and blast-like. Occasionally, well-differentiated rhabdomyoblasts may be seen as large, round, bizarre or strap cells with cross striations. These cross striations are specific for rhabdomyosarcoma, but are present only in 30% of cases.
Alveolar rhabdomyosarcoma (ARMS) is the second most common subtype seen in childhood. These tumors have either PAX3-FOXO1 or a PAX7-FOXO1 gene fusion. They commonly involve the deep soft tissue of the extremities, head and neck, paraspinal region, and perineum. On Papanicolaou (Pap) stain, cytology samples may be cellular with a mixture of reactive mesothelial cells and singly scattered, rare, bizarre, dyshesive, pleomorphic, giant tumor cells. Rhabdomyoblasts and multinucleated tumor giant cells with peripheral wreath-like nuclei, when present, are helpful diagnostic clues. Mitotic figures are easily identified. The cytoplasm of these tumor cells is eosinophilic, moderate in amount, and sometimes vacuolated. Due to their cytomorphologic variability in effusions ARMS can mimic reactive mesothelial cells and adenocarcinoma cells.27
Desmoplastic small round cell tumor (DSCRT)
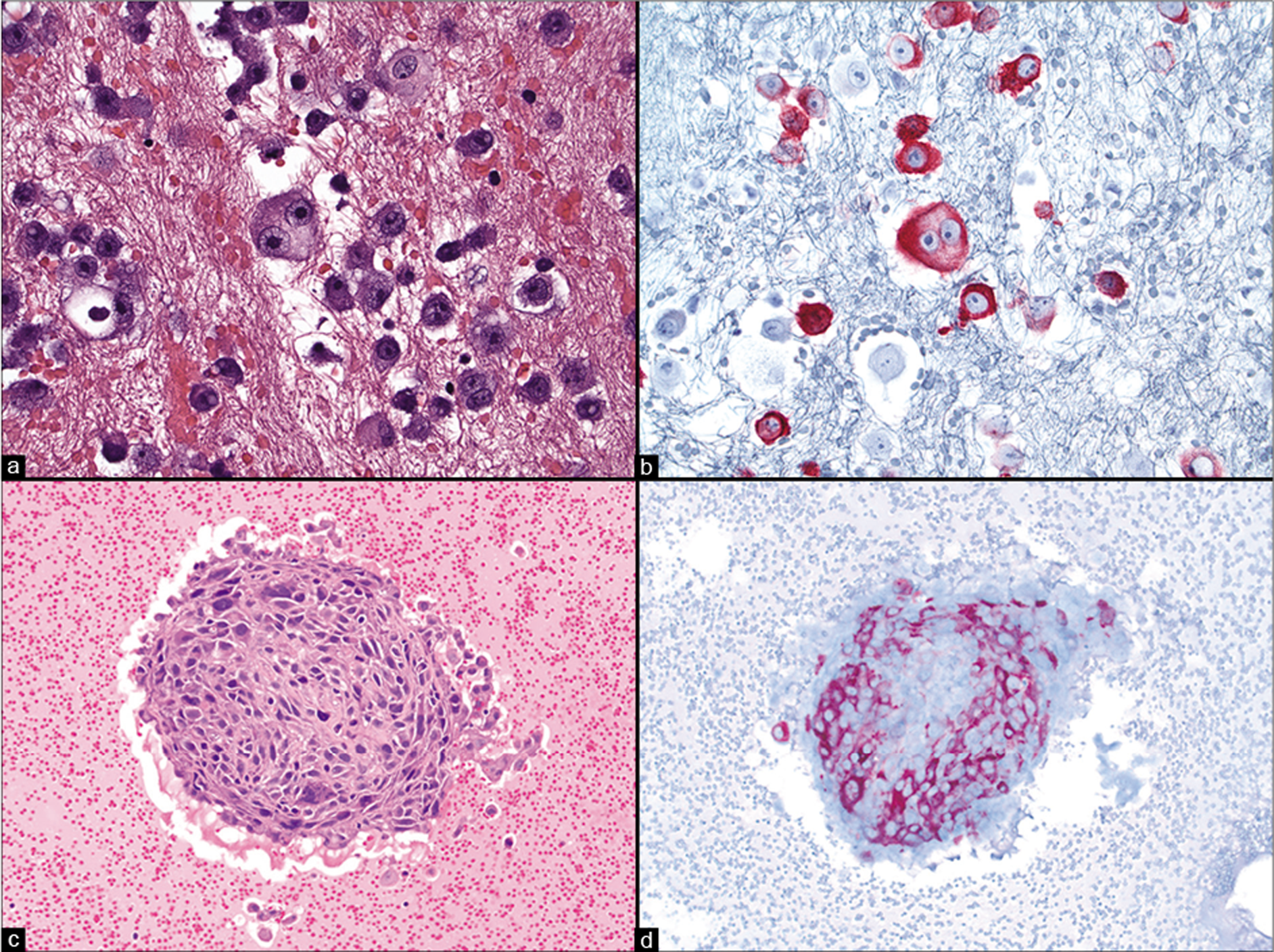
DSRCT typically involves the abdominal cavity, typically in young males, and has an aggressive course. Patients may manifest with numerous serosal implants. DSRCT of the pleura may rarely occur and present with a pleural effusion.28 This tumor is characterized by nests of proliferating small blue cells in a dense desmoplastic stroma. Effusion samples either show singly scattered tumor cells [Figure 5] or cohesive cell clusters [Figure 6] with similar cytologic features to other SRBCTs.29-30 The tumor cells tend to be uniform, have round to oval or even reniform nuclei, fine chromatin and small nucleoli. Some tumors can have clear cytoplasm and larger cells with notable anisonucleosis. There may be nuclear molding, mitoses, and apoptotic cells. Immunohistochemically, these tumors demonstrate immunoreactivity with epithelial (cytokeratin, EMA), neural (NSE, GFAP), and muscle immunomarkers (paranuclear and dot-like desmin immunostaining). WT-1 (polyclonal C-terminus antibody) is also immunoreactive in around 90% of these tumors. DSRCT has been shown to have a novel translocation t(11;22) (p13;q12), which is a chimeric fusion product of the EWSR1-WT1 gene.

-
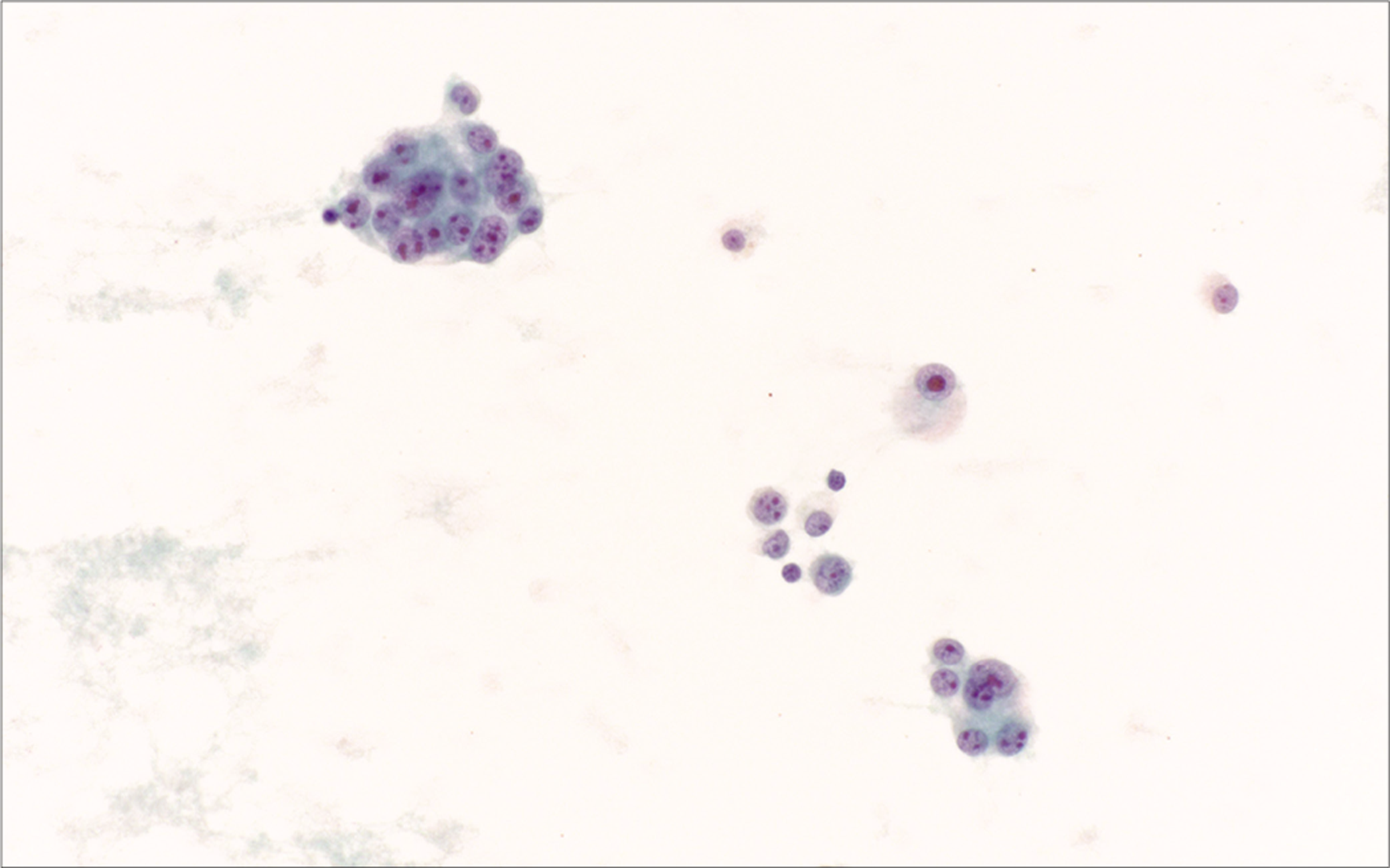
Desmoplastic small round cell tumor (DSRCT) metastatic to pleural fluid. Hypercellular sample with numerous isolated tumor cells are shown with high N:C ratios, round nuclei and finely granular chromatin. [a. Cytospin, Pap stain, 40x]

-
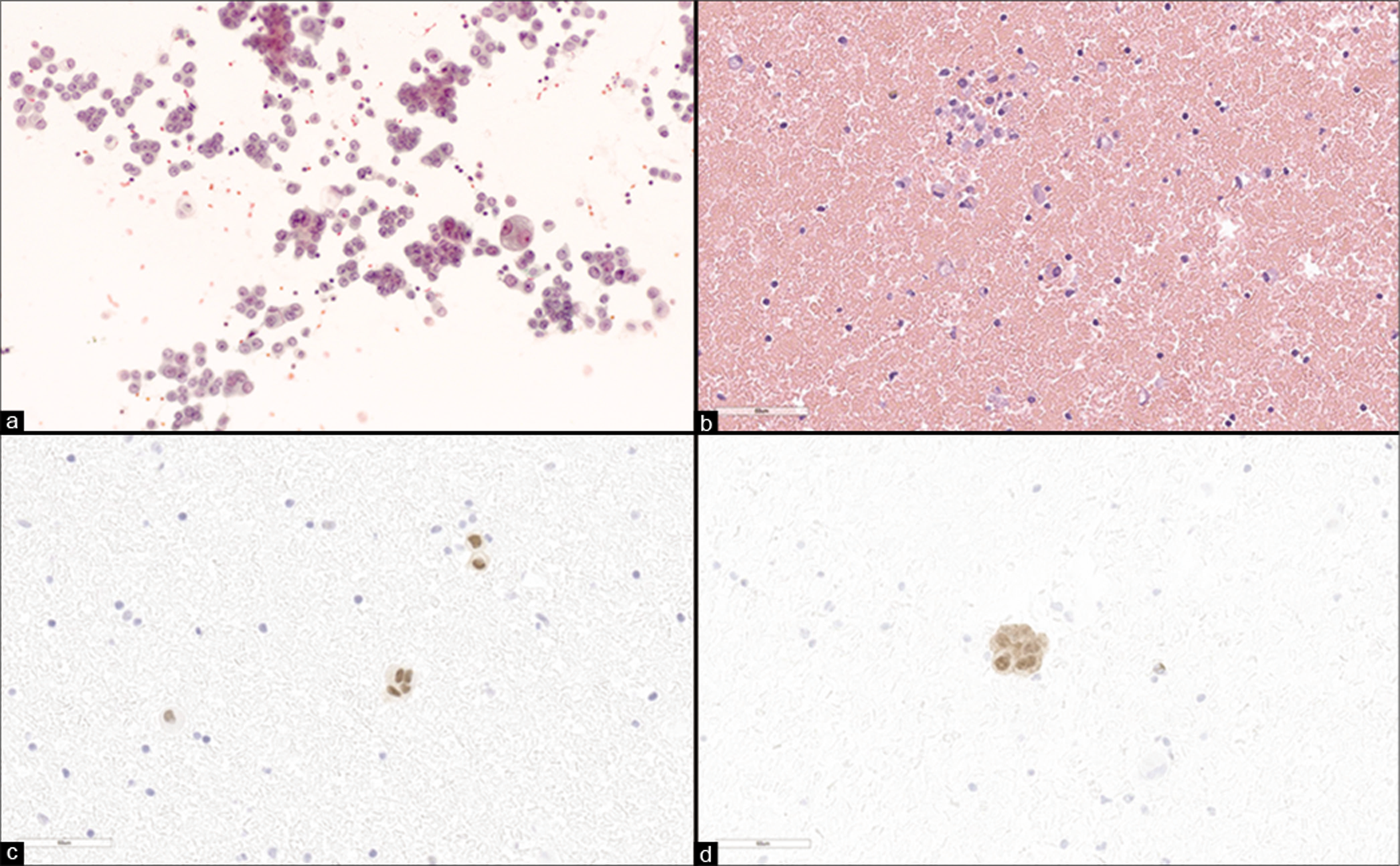
Desmoplastic small round cell tumor (DSRCT) involving peritoneal fluid. Tumor cells are shown forming a cohesive cluster (a). DSRCT tumor cells showing nuclear immunoreactivity for WT1 (arrows in b). [(a. Pap stain, 100x; b. WT-1 immunostain, 100x] (From Cytojournal 2005, 2:6. Courtesy of Granja NM et al.42)
Ewing sarcoma
The Ewing sarcoma family are sarcomas of neuroectodermal differentiation. They include Ewing sarcoma (most undifferentiated), peripheral primitive neuroectodermal tumor (PNET) and Askin tumor of the chest wall. They are the second most common sarcoma of bone (after osteosarcoma) and soft tissue (after rhabdomyosarcoma) in children and young adults. Visceral involvement can also occur. Primary extraskeletal Ewing sarcoma of the pleura is rare.31 Approximately 25% of afflicted patients may present with metastases at the time of diagnosis and spread is preferentially to the lungs, bone marrow, and lymph nodes.
Metastases involving pleural effusions are infrequent and found in less than 10% of cases.32 A malignant effusion may be the primary presentation in some patients.33 Effusions involved by Ewing sarcoma may show loose groups of tumor cells or many solitary cells. These cells have a high nuclear-cytoplasmic ratio characterized by oval nuclei with scant, ill-defined cytoplasm [Figure 7]. Their chromatin is finely granular, and nucleoli are small and indistinct. Apoptosis and necrosis may be evident.

-
Ewing sarcoma metastatic to pleural fluid. This loose cluster is comprised of small round tumor cells with minimal cytoplasm, round to oval nuclei, finely granular chromatin and distinct nucleoli. [a. Smear, Pap stain, 60x]
The tumor cells contain intracytoplasmic glycogen instead of mucin, which can be demonstrated with a positive periodic acid-Schiff (PAS) stain. The typical tigroid background due to smeared glycogen seen on stained slides is missing in effusions. These tumors can be differentiated from other SRBCTs using ancillary studies. There is often strong and diffuse membranous immunoexpression of CD99 (antibody to MIC2) and diffuse NKX2.2 staining. However, CD99 is also immunoexpressed in most of the other SRBCTs except for neuroblastoma and so may be relatively non-specific. Almost 25% of cases may express keratin and rarely there may be desmin immunoreactivity. Most tumors demonstrate t(11;22) EWSR1-FLI1 (85%) or t(21;22) EWSR1-ERG (10%) gene fusions. Detecting transcripts may be necessary to clinch the diagnosis.
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor encountered in children. Neuroblastoma and related tumors are derived from primordial neural crest cells that migrate from the mantle layer of the spinal cord and populate the primordia of the sympathetic ganglia and adrenal medulla. The age of presentation ranges from 2 months to 5 years, with a peak incidence at 18 months. The distribution of neuroblastomas follows the sympathetic chain and can arise anywhere from the base of skull to pelvis, including the adrenal medulla and dorsal root ganglia. About 90% of neuroblastomas are associated with elevated levels of catecholamines. Measurement of these metabolites in the urine is useful in monitoring the course of disease. The incidence of pulmonary involvement at the time of diagnosis is low, averaging about 4% in children over 1 year old with stage IV neuroblastoma.
In a report by Cowie et al, the incidence of pleural disease or pleural effusion in a group of 1,245 patients with neuroblastoma was 0.7%.34 Pleural effusions usually signify a neuroblastoma with unfavorable biologic features and high-risk disease.35 Cytologically, neuroblastomas are composed of small blue tumor cells [Figure 8]. These cells may form Homer-Wright rosettes, that can rarely be observed in effusions. The cytoplasm of neuroblastoma cells is scant and their nuclei have salt and pepper-type chromatin. Ganglion cells seen with maturation are unlikely to be encountered among metastatic tumor cells to body fluids. Neuroblastoma cells are variably immunoreactive for synaptophysin and chromogranin, and may also immunostain with GFAP, GATA3, tyrosine hydroxylase, and PHOX2B. A helpful immunopanel to distinguish neuroblastoma from other small blue cell tumors includes neuron specific enolase (NSE) and protein gene product 9.5 (PGP 9.5). However, these immunostains are relatively non-specific. Flow cytometry has proven to be helpful, for the detection of CD56+ neuroblastoma cells.36 Several cytogenetic abnormalities have been reported, some of which are associated with a poor prognosis (e.g. ATRX, MYCN, and ALK amplifications).

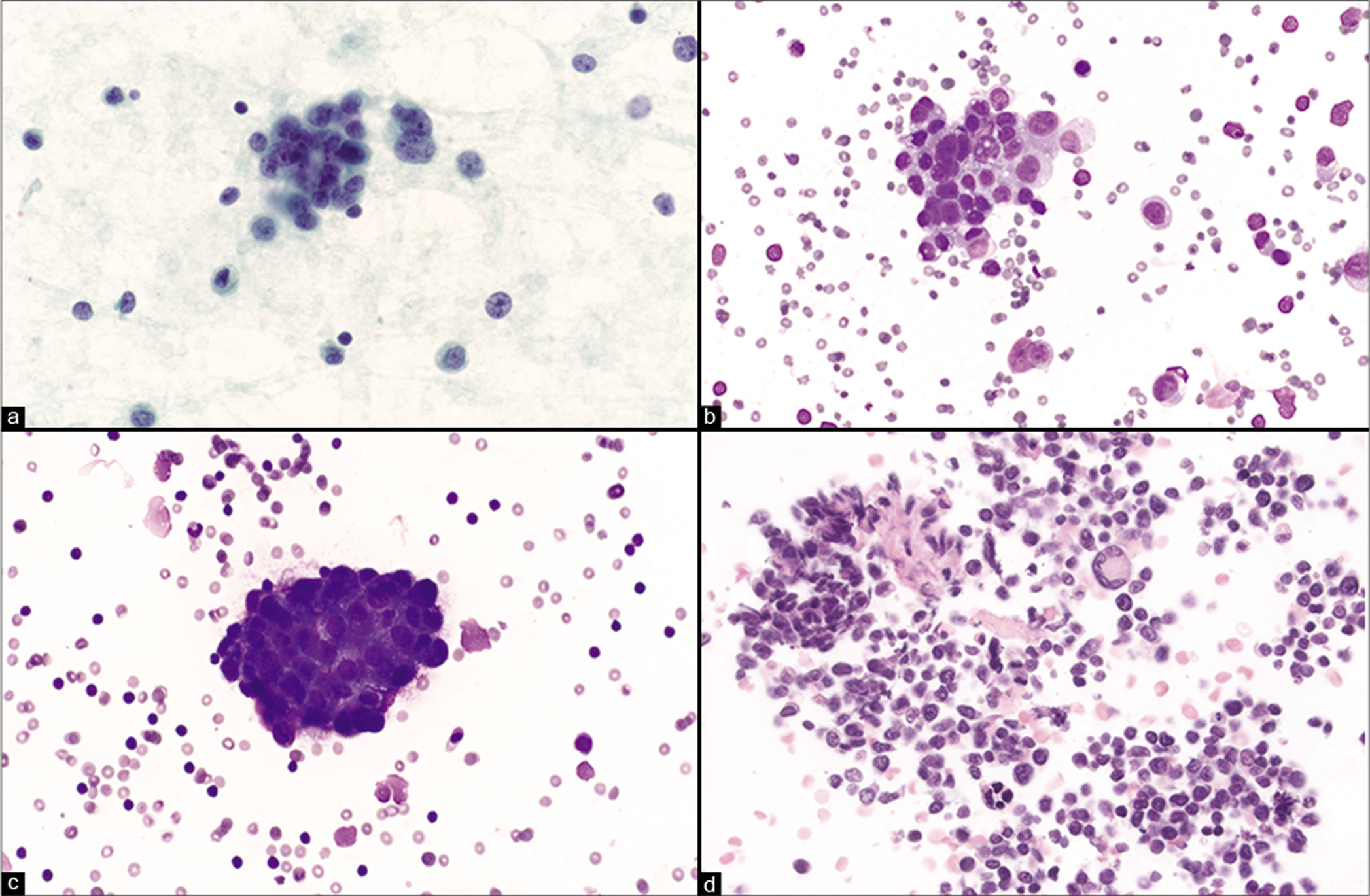
-
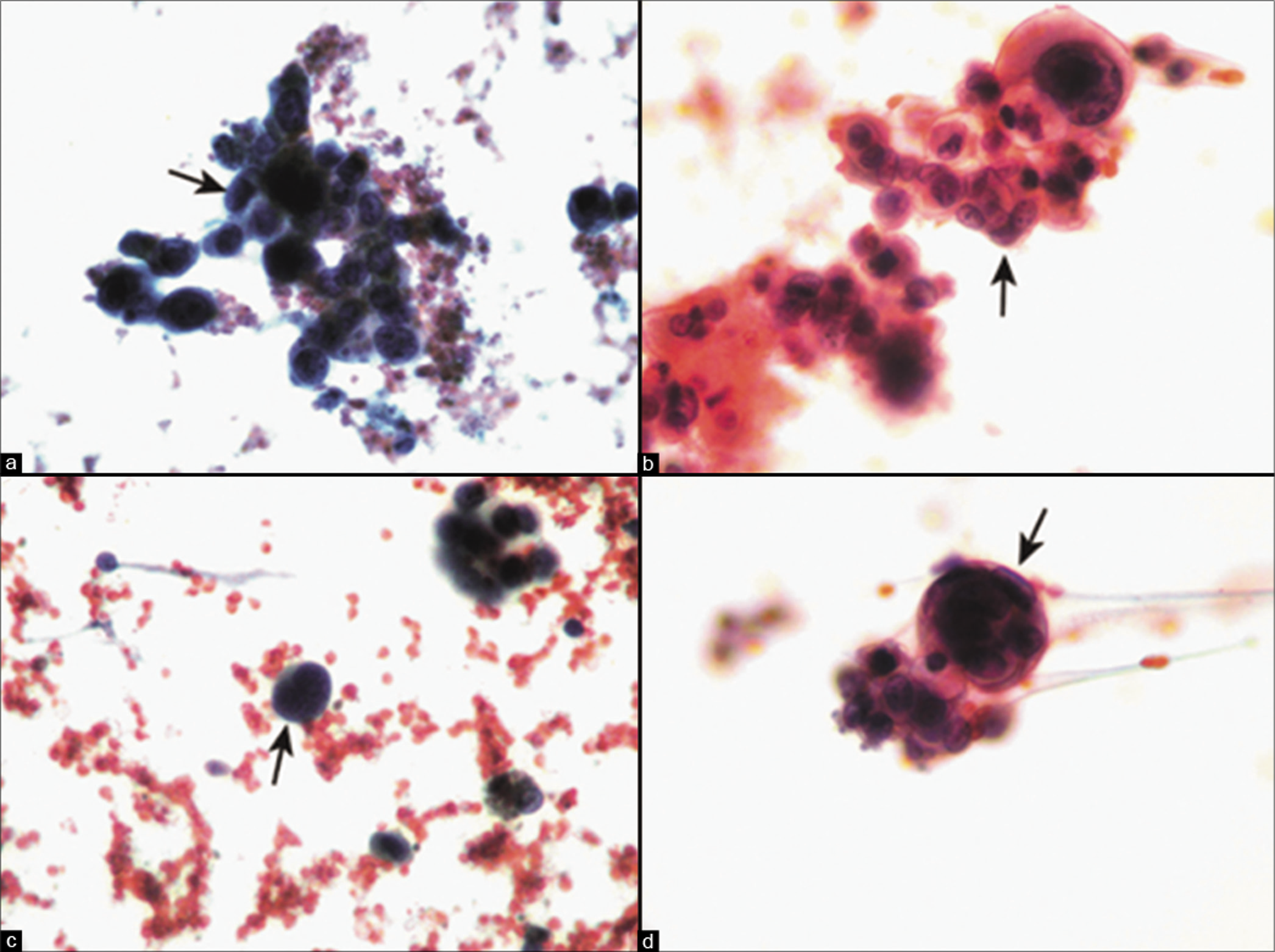
Neuroblastoma metastatic to peritoneal fluid. Tumor cells shown forming a cohesive cluster (a). Hypercellular bloody fluid sample with many small round blue tumor cells (b). [a. Smear, DQ stain, 60x; b. Cell-block, H&E stain, 40x]
Wilms tumor
Wilms tumor (WT) or nephroblastoma, a pediatric renal malignancy, is the most common solid tumor in children. Relapse usually occurs within 4 years of initial presentation, and the most common site of metastasis is the lung. Pleural effusion at the time of presentation is rare and is not associated with an adverse prognosis.37 Occasionally, a Wilms tumor can rupture spilling tumor cells into the abdominal cavity. Histologically, WT is heterogeneous with epithelial, stromal, and blastemal (undifferentiated) components. When there is a predominance of one of these components, this will make the diagnosis challenging, especially in a metastatic setting. The cytology of WT in effusions has been rarely reported in the literature.38 Tumor cells present mostly as cellular aggregates [Figure 9]. Blastema consists of small blue tumor cells. More complex cystic and tubular structures may be noted when epithelial components are present. Immunohistochemistry is helpful to distinguish these tumors from other SRBCTs. Strong immunoreactivity for WT-1 is typically demonstrated in epithelial and blastemal components, but not stromal components. Well-differentiated blastemal components may exhibit cytokeratin immunoreactivity. Molecular aberration of chromosome 11p, where the WT-1 gene is located, may be useful in establishing the diagnosis.

-
Wilms tumor metastatic to peritoneal fluid. Tumor cells are shown with variable N:C ratios, round nuclei and uniform chromatin. [Smear, DQ stain, 60x]
Other sarcomas
Almost any sarcoma can metastasize to involve serous effusions [Figures 10 and 11].39-41 A diagnosis of metastatic sarcoma to serous fluid is typically rendered in the setting of a known primary tumor. In a small series with 24 cases of malignant effusions secondary to sarcomas there were a variety of tumor types (e.g. leiomyosarcoma, rhabdomyosarcoma, liposarcoma, synovial sarcoma, osteosarcoma, chondrosarcoma, and high-grade sarcoma NOS).8 The associated cytomorphology for this series included single cell arrangement of tumor cells in 96% of cases, indistinct cell borders in 75%, nuclear pleomorphism in 75%, multinucleation in 54%, and a proteinaceous background with lysed blood in 71%. Epithelioid hemangioendothelioma (EHE) involving serous effusions is not only characterized by neoplastic epithelioid to plasmacytoid cells present singly or in small clusters, but also by the presence of intracytoplasmic erythrocytes [Figure 12].42 EHE can be confirmed by immunoreactivity of tumor cells to vascular immunomarkers (CD31, CD34, ERG) with nuclear immunoexpression for CAMATA1. Characterization of a sarcoma involving serous fluids in many cases is only possible with adequate clinical history, comparative evaluation of the primary sarcoma, and ancillary studies. It is important to be aware that these sarcomas may exhibit a variety of features that can differ from those of the original neoplasm and may thus preclude the correct diagnosis. With Kaposi sarcoma, for example, bizarre amoeboid cells have been reported in pleural effusions.43
- Metastatic fibrosarcoma to peritoneal fluid. Poorly cellular specimen with predominantly scattered single neoplastic cells (arrows). The sarcoma cells have high N/C ratio with moderate amount of cytoplasm and irregular nuclei with frequent binucleation and multinucleation. [a– c, Pap stain, 10x]
-
Clear cell sarcoma metastatic to pleural fluid. Epithelioid tumor cells have delicate cytoplasm and round nuclei with prominent nucleoli. [Smear, Pap stain, 40x]
- Pleural fluid involved by metastatic epithelioid hemangioendothelioma (EHE). Hypercellular sample with many epithelioid tumor cells (a). Bloody cell-block showing occasional tumor cells with binucleation (“bug-eyed demons”) (b). Tumor cells show nuclear ERG immunoreactivity (c). Tumor cells are immunoreactive for CAMTA1 (d). [a. Smear, Pap stain, 20x; b. cell-block, H&E stain, 10x equivalent; c. cell-block, IHC stain, 20x equivalent; d. cell-block, IHC stain, 20x equivalent]
MALIGNANT MELANOMA
Malignant melanomas are typically seen in people with fair complexion in sun-exposed areas of skin such as the head, neck, and lower extremities. In recent years, the incidence of malignant melanoma has increased; however, effusions due to melanoma are relatively rare and constitute only 2–3% of malignant effusions.44,45 Metastatic melanoma may cause effusion samples to appear black.46 A black pleural effusion can also be caused by fungal infection (Aspergillus niger or Rhizopus oryzae), pancreaticopleural fistula, and hemolysis after massive intrapleural bleeding.
Cytomorphology
The morphologic diagnosis of malignant melanoma in effusion cytology specimens may be difficult to make due to the wide variety of cellular patterns that may be seen. Melanoma usually demonstrates striking cellular pleomorphism, with predominantly dyshesive epithelioid cells. Malignant melanoma is also notorious for presenting with a wide spectrum of cytomorphology.47,48 Several variants have been described including spindle cell, small cell, balloon cell, granular cell, and signet-ring cell variants. Melanoma cells may accordingly be epithelioid, spindle-shaped, or extremely bizarre and resemble carcinoma, sarcoma, or anaplastic lymphoma. This is further complicated because of changes secondary to physical forces, such as the surface tension phenomenon associated with fluids.
Malignant melanoma cells in effusions usually present as singly scattered cells, but cell cluster formation may be seen [Figure 13]. Most of the samples contain blood and various inflammatory components in the background with reactive mesothelial cells and scattered histiocytes. Cell-in-cell pattern may be present. Malignant melanoma cells that are round to oval in shape can show extreme variability in size, ranging from one to five times the size of mesothelial cells. The cytoplasm of these tumor cells is invariably abundant, causing a surprisingly low nuclear-cytoplasmic ratio (N:C ratio). Cells can be vacuolated, with evenly distributed large and small vacuoles. Signet-ring cytoplasmic vacuolization may also rarely be seen. DQ-stained tumor cells may exhibit a two-zone staining pattern with inner, darker, basophilic staining and an outer rim of magenta cytoplasm resembling reactive mesothelial cells. The presence of cytoplasmic melanin pigment is variable, and has been reported in approximately 50–83% of cases. The tumor cells in effusions may be heavily or minimally pigmented [Figure 14], or even amelanotic. Interestingly, primary amelanotic or minimally pigmented melanomas may give rise to heavily pigmented tumor cells in effusions and vice versa. Melanin pigment appears as fine, black to dark blue cytoplasmic granules in DQ-stained material. In Pap-stained preparations, melanin pigment appears as non-refractile fine brown-black cytoplasmic granules, which can condense into coarser granules that may resemble hemosiderin. A Fontana–Masson stain may be used to confirm melanin pigment.
- Metastatic melanoma to peritoneal fluid. The malignant cells may be present in clusters (arrows in a, b) but are usually dyshesive (arrow in c). Signet-ring type cytoplasmic vacuoles and ‘cell-in-cell’ pattern (arrow in d) may be present. [a–d, Pap stain, a-d. 40x]
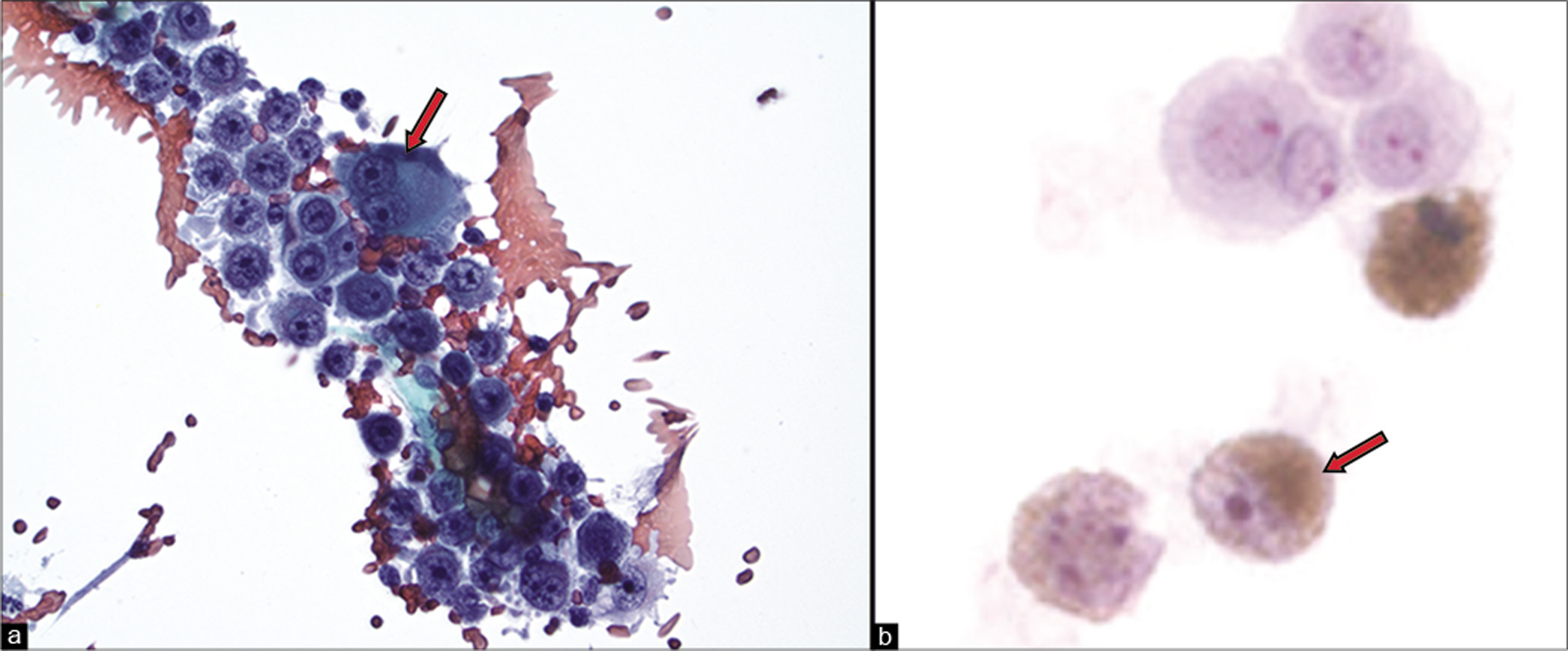
- Metastatic melanoma to peritoneal fluid. Epithelioid tumor cells have large round nuclei with prominent nucleoli and occasional cells with binucleation (“bug-eyed demons”) (arrow) (a). Melanoma cells with fine brown melanin pigment (arrow) (b). [a. Cytospin, Pap stain, 40x; b. ThinPrep, Pap stain, 40x]
The large, round to oval nuclei of malignant cells are usually eccentrically placed (plasmacytoid cells). Only rare malignant cells may show central nuclei and/or irregular nuclear contours. Their chromatin pattern is usually finely granular or lacy; however, coarse aggregations may be noted. In general, the nuclei of melanoma cells are hyperchromatic, containing a single or multiple prominent nucleoli. Macronucleoli are a typical nuclear feature [Figure 15a], but are not always present. Binucleated cells (“bug-eyed demons”) are characteristically present and multinucleated cells can also be seen. Cytoplasmic intranuclear pseudoinclusions are present in some cases and are a useful feature. Mitoses may be rare to absent.
- Metastatic melanoma to body fluids confirmed by immunoreactivity to melanocytic immunomarkers in cell-block material. Dyshesive tumor cells with frequently observed binucleated cells (“bug-eyed demons”) (a) are immunoreactive for Melan-A (b). Many of the melanoma cells within this tightly cohesive pleural fluid cluster (c) are Melan-A immunoreactive (red chromogen) (d). [a & c. H&E stain; b & d. IHC stain; a & b. 40x; c & d. 20x]
Differential diagnosis
Cases without pigment may be difficult to differentiate from other neoplasms (e.g. lymphoma) involving the serous cavities. Immunostaining for leukocyte common antigen (LCA, CD45) favors lymphoma. Large binucleated melanoma cells with prominent nucleoli may suggest Reed– Sternberg cells of Hodgkin disease. However, in contrast to Reed–Sternberg cells, melanoma cells do not express CD15 (Leu MI) or CD3O (Ber-H2). Some cases of melanoma may show a signet-ring-cell appearance and hence could resemble signet-ring cell carcinoma. However, signet-ring carcinomas typically contain mucin within their cytoplasmic vacuole, a feature not associated with melanoma. Although uncommon, melanoma cells with balloon cell or granular cell features may be confused with renal cell carcinoma, hepatocellular carcinoma, or other clear cell tumors.
One of the pitfalls is interpreting hemosiderin-laden macrophages associated with chronic hemorrhagic effusions as melanin-containing melanoma cells. However, in most cases, the bland nuclear features of macrophages with brown pigment (either siderophages with hemosiderin or melanophages with melanin) help to distinguish them from melanoma cells. Classic single, large, multinucleated cells with prominent nucleoli, intranuclear cytoplasmic pseudoinclusions, and abundant melanin pigment generally do not pose a diagnostic challenge. However, these cellular features are represented in a minority of cases. Immunostaining for melanoma immunomarkers is thus helpful to confirm the diagnosis in most cases.
Immunocytochemistry
The ancillary technique that has proved to be most practical in the identification of melanoma is immunocytochemistry. Melanoma tumor cells may be immunoreactive for S-100 protein, HMB45, and other melanoma immunomarkers including SOX-10, Melan-A/Mart-1, tyrosinase, MITF, and PRAME. Malignant melanoma cells are immunoreactive for Melan-A/Mart-1 in 80% of melanomas [Figure 15]. Vimentin, while positive in melanomas, is also positive in mesotheliomas, sarcomas, lymphoma, and some carcinomas. Cytokeratin immunoreactivity has been reported in malignant melanoma in 1–27% of cases, but such aberrant immunostaining in these cells is usually weak and focal.
GERM CELL NEOPLASMS
Germ cell neoplasms are common in pediatric and young adults. They may arise from the gonads, but could also occur in other extra-gonadal locations including the abdomen, mediastinum, and within the brain. Based on their pathology, they can be classified as follows: teratomas (benign and malignant), seminoma (in males) or dysgerminoma (in females), yolk sac tumor (endodermal sinus tumor), embryonal carcinoma, and choriocarcinoma. Mixed tumors with varying components may occur. Advanced neoplasms can spread to the lung, liver, and other sites, and may manifest with malignant effusions. In body fluids, the cytomorphology of a germ cell tumor may be similar to or differ from the original neoplasm. Large mediastinal germ cell tumors may also be associated with a reactive pleural effusion.49
While benign teratomas (e.g. ovarian dermoid cyst) typically do not metastasize, these tumors may be encountered in effusion cytology samples when pelvic washings accompany their surgical removal and/or cyst rupture. Spillage is more likely to be seen with the trend toward employing laparoscopy to manage benign ovarian masses. In this setting, spilled keratin material can evoke a marked granulomatous response (chemical or granulomatous peritonitis).50 Immature teratomas have the potential to metastasize. The cytologic examination of ascitic fluid in such cases may reveal a variety of neoplastic cells, including immature neuroepithelial cells forming rosette-like structures, keratinized squamous cells, squamoid metaplastic cells, and immature glial-appearing cells.51
Metastatic seminoma can form loose groups or single cells. These tumor cells are often larger than mesothelial cells. They are uniform in size with prominent nucleoli. Mitotic figures are frequent. The usual lymphocytes and frothy background seen with aspirated seminomas may not be present in effusions.
Yolk sac tumor in ascites shows small tight clusters or rare tubulovesicular structures. The uniform cells of yolk sac tumor may sometimes show intracytoplasmic hyaline droplets.52 The variable cytomorphology of yolk sac tumor (e.g. round cells with multiple prominent nucleoli and vacuolated or mucin-containing cytoplasm) may result in the misinterpretation as adenocarcinoma.
Embryonal carcinoma has large, pleomorphic cells with prominent nucleoli and numerous mitoses. Such cytology may mimic a poorly differentiated adenocarcinoma.
Choriocarcinoma is a biphasic tumor composed of syncytiotrophoblasts and cytotrophoblasts. These two components, however, are usually not seen in effusions together. In effusions, syncytiotrophoblasts occur singly as large, multinucleated cells. The cytotrophoblasts can present in loose groups or as singly distributed basophilic, small cells with small eccentric nuclei. These cells may be difficult to distinguish from metastatic adenocarcinoma. Cell-blocks are particularly useful for performing immunohistochemical studies to highlight and differentiate different components of mixed germ cell tumors.
Acknowledgment
The authors of this review and editors of CMAS #2 series thank the initial authors (Mamatha Chivukula, MD and Reda Saad, MD) for all their efforts for the first edition material on which the current review (as chapter #10 in the final CMAS #2) is based.
The authors thank Andrew Kumar, MD (Resident, Wayne State University School of Medicine, Detroit, MI, USA) and Janavi Kolpekwar for copy-editing support.
ABBREVIATIONS (IN ALPHABETIC ORDER)
ARMS = Alveolar rhabdomyosarcoma
DQ = Diff-Quik
DSRCT = Desmoplastic small round cell tumor
EHE = Epithelioid hemangioendothelioma
MMMT = Malignant mixed Müllerian tumors
MPNST = Malignant peripheral nerve sheath tumor
N:C ratio = Nuclear-cytoplasmic ratio
Pap = Papanicolaou
PAS = Periodic acid-Schiff
PNET = Peripheral primitive neuroectodermal tumor
RMS = Rhabdomyosarcoma
SRBCT = Small round blue cell tumor
References
- Cytologic diagnosis of a metastatic oligodendroglioma in a pleural effusion. A case report. Acta Cytol. 2006;50:542-4.
- [CrossRef] [PubMed] [Google Scholar]
- Cytological diagnosis of metastatic glioblastoma in the pleural effusion of a lung transplant patient. Diag Cytopathol. 2014;42:619-23.
- [CrossRef] [PubMed] [Google Scholar]
- Extracranial glioblastoma diagnosed by examination of pleural effusion using the cell block technique: case report. Neurosurg Focus. 2018;44:1-5.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse midline glioma metastasis to the periotoneal cavity via ventriculo-peritoneal shunt: Case report and review of literature. J Clin Neurosci. 2019;67:288-93.
- [CrossRef] [PubMed] [Google Scholar]
- Cytologic and immunophenotypic features of malignant cells in pediatric body fluids. Acta Cytol. 2015;59:332-8.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical, cytologic, and immnohistochemical features of sarcomas involving body cavity fluids. Cancer Cytopathol. 2019;127:778-84.
- [CrossRef] [PubMed] [Google Scholar]
- Bone and soft tissue sarcomas in cerebrospinal fluid and effusion: a 20-year review at our institution. Cancer Cytopathol. 2021;129:776-87.
- [CrossRef] [PubMed] [Google Scholar]
- Fine needle aspiration of primary soft tissue tumors. Morphologic analysis of the most frequent types. Acta Cytol. 1992;36:905-17.
- [Google Scholar]
- CellBlockistry: Chemistry and art of cell-block making-A detailed review of various historical options with recent advances (Review) CytoJournal. 2019;16:12. (28 June 2019) Available FREE in open access from: http://alturl.com/kn8p4
- [CrossRef] [PubMed] [Google Scholar]
- 2021. ISBN Print version 978-1-955571-00-5; e-Version 978-1-955571-01-2 https://www.amazon.com/-/es/Vinod-Shidham-ebook/dp/B098B65PSL This book is available FREE under eCytoJournal at: https://cytojournal.com/eissues/
- [Google Scholar]
- Architectural aspects of cell-blocks as small biopsies. Cytojournal. 2021;18:5.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- CellBlockistry: Science of Cell-Block Making as Ancillary Cytopathology Component in the Era of Minimally Invasive Techniques with Increasing Role of Molecular Pathology (Invited Short communication) Clinics in Surgery 2019 Volume 4; Article 2510 Remedy Publications LLC. http://clinicsinsurgery.com/ Available FREE at: http://www.clinicsinsurgery.com/pdfs_folder/cis-v4-id2510.pdf
- [Google Scholar]
- Peritoneal washing cytology findings of disseminated myxoid leiomyosarcoma of uterus: report of a case with emphasis on possible differential diagnosis. Diagn Cytopathol. 2002;27:47-52.
- [CrossRef] [PubMed] [Google Scholar]
- Cell-blocks and other ancillary studies (including molecular genetic tests and proteomics) Cytojournal. 2021;18:4.
- [CrossRef] [PubMed] [Google Scholar]
- Immunocytochemical panel for the identification of malignant cells in serous effusions. Am J Clin Pathol. 1991;95:867-74.
- [CrossRef] [PubMed] [Google Scholar]
- Effusion cytomorphology of small round cell tumors. J Cytol. 2016;33:85-92.
- [CrossRef] [PubMed] [Google Scholar]
- A review of effusion cytomorphology of small round cell tumors. Acta Cytol 2021 In press
- [CrossRef] [PubMed] [Google Scholar]
- Cytologic diagnosis of rhabdomyosarcoma in a patient with germ cell tumor. A case report. Acta Cytol. 1995;39:249-51.
- [Google Scholar]
- Prognostic role of pleural effusion or ascites in localized rhabdomyosarcoma. Pediatr Blood Cancer. 2019;66:e27932.
- [CrossRef] [PubMed] [Google Scholar]
- Rhabdomyosarcoma in a child with massive pleural effusion. Cytologic diagnosis from pleural fluid. Diag Cytopathol. 1999;21:125-8.
- [CrossRef] [Google Scholar]
- Cytological diagnosis of rhabdomyosarcoma in a child with a pleural effusion. A case report. Acta Cytol. 2004;48:249-53.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of alveolar rhabdomyosarcoma in effusion cytology: a diagnostic pitfall. Cytopathology. 2010;21:273-5.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural effusion cytology of embryonal rhabdomyosarcoma. Diag Cytopathol. 1997;16:270-3.
- [CrossRef] [Google Scholar]
- Alveolar rhabdomyosarcomas involving serous cavity fluid specimens exhibit diverse cytomorphologies: A case report and review of the literature. Diagn Cytopathol. 2020;48:1155-61.
- [CrossRef] [PubMed] [Google Scholar]
- A rare cause of bilateral pleural effusion-desmoplastic small round cell tumor. Monaldi Arch Chest Dis 2021 in press
- [CrossRef] [PubMed] [Google Scholar]
- Effusion cytology of desmoplastic small round cell tumor of the pleura. A case report. Acta Cytol. 1993;37:77-82.
- [Google Scholar]
- Desmoplastic small round cell tumour: cytological and immunocytochemical features. Cytojournal. 2005;2:6.
- [CrossRef] [PubMed] [Google Scholar]
- A primary Ewing's sarcoma of pleura: Case report and literature review. Respir Med Case Rep. 2021;34:101516.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural effusion in a patient with Ewing sarcoma. Cytopathology. 2022;33:138-40.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural Effusion Revealing a Diagnosis of Ewing Sarcoma. Cureus. 2021;13:e20439.
- [CrossRef] [Google Scholar]
- Lung involvement in neuroblastoma: incidence and characteristics. Med Pediatr Oncol. 1997;28:429-32.
- [CrossRef] [Google Scholar]
- Significance of pleural effusion in neuroblastoma. Pediatr Blood Cancer. 2007;49:906-8.
- [CrossRef] [PubMed] [Google Scholar]
- Comparative Analysis of Flow Cytometry and Cytomorphology for Neuroblastoma Cell Detection in Effusion and Bone Marrow Specimens. Fetal Pediatr Pathol. 2019;38:1-7.
- [CrossRef] [PubMed] [Google Scholar]
- Significance of pleural effusion at diagnosis of Wilms' tumor. Pediatr Blood Cancer. 2004;42:145-8.
- [CrossRef] [PubMed] [Google Scholar]
- Cytological identification of metastatic epithelial nephroblastoma in pleural fluid: report of a case and review of literature. Diagn Cytopathol. 2006;34:621-5.
- [CrossRef] [PubMed] [Google Scholar]
- Cytology of synovial sarcoma metastases in pleural fluid. Acta Cytol. 1982;26:517-20.
- [Google Scholar]
- Exfoliative cytology of clear-cell sarcoma metastases in pleural fluid. Diagn Cytopathol. 1986;2:144-9.
- [CrossRef] [PubMed] [Google Scholar]
- Primary ovarian angiosarcoma presenting as malignant cells in ascites: case report and review of the literature. Diagn Cytopathol. 2005;32:307-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cytologic features and immunohistochemical findings of epithelioid hemangioendothelioma (EHE) in effusion: a case series. Diag Cytopathol. 2021;49:E24-E30.
- [CrossRef] [PubMed] [Google Scholar]
- The detection of acquired immunodeficiency syndrome-associated Kaposi sarcoma cells in pleural effusion by CD34 immunostain. Cancer. 1993;72:2260-5.
- [CrossRef] [Google Scholar]
- Malignant melanoma presenting as an isolated pleural effusion. Monaldi Arch Chest Dis. 2011;75:138-40.
- [CrossRef] [PubMed] [Google Scholar]
- Pleural metastasis from auricular melanoma: A brief report. Diagn Cytopathol. 2020;48:376-9.
- [CrossRef] [PubMed] [Google Scholar]
- Black pleural effusion: an unusual presentation of metastatic melanoma diagnosed by medical thoracoscopy. Respirol Case Rep. 2019;7:e00490.
- [CrossRef] [PubMed] [Google Scholar]
- Effusion cytomorphology and immunocytochemistry of malignant melanoma: five cases of melanotic melanoma and one case of amelanotic melanoma. Diagn Cytopathol. 2009;37:516-21.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic clue for pleural metastasis of malignant melanoma. J Gen Fam Med. 2018;19:217-8.
- [CrossRef] [PubMed] [Google Scholar]
- Germ cell tumor causing pleural effusion: A diagnostic dilemma. Indian J Tuberc. 2017;65:80-3.
- [CrossRef] [PubMed] [Google Scholar]
- Chemical peritonitis: a rare complication of an iatrogenic ovarian dermoid cyst rupture. Surg Endosc. 2003;17:658.
- [CrossRef] [PubMed] [Google Scholar]
- Cytomorphologic features of immature ovarian teratoma in peritoneal effusion: a case report. Diagn Cytopathol. 2005;33:39-42.
- [CrossRef] [PubMed] [Google Scholar]
- Endodermal sinus tumor of the mediastinum. Cytologic diagnosis on a pleural effusion. Acta Cytol. 1990;34:257-60.
- [Google Scholar]




